
Constructing Genetic Linkage Maps Using the Whole Genome Sequence of Pacific Bluefin Tuna (Thunnus o
Vol.07No.02(2016), Article ID:63702,38 pages
10.4236/abb.2016.72010
Tsubasa Uchino1, Yoji Nakamura2, Masashi Sekino2, Wataru Kai2, Atushi Fujiwara2, Motoshige Yasuike2, Takuma Sugaya3, Himeko Fukuda1, Motohiko Sano1, Takashi Sakamoto1*
1Faculty of Marine Science, Tokyo University of Marine Science and Technology, Tokyo, Japan
2Research Center for Aquatic Genomics, National Research Institute of Fisheries Science, Fisheries Research Agency, Yokohama, Japan
3Research Center for Marine Invertebrate Animals, National Research Institute of Fisheries and Environment of Inland Sea, Fisheries Research Agency, Hiroshima, Japan 
Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 January 2016; accepted 21 February 2016; published 24 February 2016
ABSTRACT
Pacific bluefin tuna (Thunnus orientalis) is one of the most economically important species in the Percomorpha group of teleost fishes. Their migrations are extensive and depend upon continuous swimming at a high rate of speed throughout their life. The draft genome sequence of this species has been reported but remains highly fragmented. We constructed a Pacific bluefin tuna genetic linkage map using microsatellite markers developed on each of the scaffolds from the draft genome sequence to link these genome fragments and understand the genomic structure of species in Percomorpha. Of the 606 polymerase chain reaction microsatellite primer pairs tested, 473 were polymorphic in the mapping populations for the linkage analysis. We constructed sex-spe- cific maps for 24 linkage groups consisting of 470 markers, which allowed us to place scaffolds that cumulatively represented 20.8% (153.8 Mb) of the sequenced genome onto the linkage groups. The distribution of orthologous genes on the chromosomes of tuna and four other teleost fish species suggested that the constitution of tuna chromosomes is closest to that of medaka. Both species have the 24 chromosomes of the ancestral teleost, including several chromosomal inversions. The integrated map developed in this study will be useful to construct a complete physical map to conduct comparative teleost genomics and genetic studies on economically useful traits in Pacific bluefin tuna.
Keywords:
Pacific Bluefin Tuna, Microsatellite Marker, Genetic Linkage Map

1. Introduction
Pacific bluefin tuna (Thunnus orientalis) is one of the most economically important species of fish, and their migrations are unique, as they swim quickly and continuously throughout their life. This continuous swimming ability enables Pacific bluefin tuna to migrate long distances in the Pacific Ocean. Continual swimming is required, so that water containing oxygen flows continuously over the gills; this is a special feature of Thunnus and closely related species. Tuna have superior swimming ability due to a large quantity of red muscle, a rapid metabolic rate, large body size, and their unique shape and swimming form, although details remain unknown. Moreover, the tuna growth rate is very high, as they can grow 50 cm in total length in 1 year. Maximum length and weight are about 2.5 m and 300 kg, respectively. These unique ecological features of Pacific bluefin tuna originate from their genome.
Tuna farmers generally use natural seed for broodstock. However, tuna catch is restricted by international fishing regulations created as a result of recent decreases in wild population numbers [1] . Thus, a breeding system that depends on the recruitment of naturally occurring siblings is difficult to maintain. The complete lifecycle has been established recently for Pacific bluefin tuna aquaculture [2] and artificial seed is now being used for production. Therefore, there is much interest in creating broodstock with commercially valuable genetic traits.
Several whole genome sequences have been reported recently in teleosts because of the development of high-throughput sequencing methods. Whole genome sequences have been registered in public databases for model fish, such as zebrafish (Danio rerio) [3] and medaka (Oryzias latipes) [4] as well as for other fish, such as fugu (Takifugu rubripes) [5] , Tetraodon (Tetraodon nigroviridis) [6] , stickleback (Gasterosteus aculeatus) [7] , and Pacific bluefin tuna [8] .
The draft Pacific bluefin tuna genome sequence was generated using the Roche 454 FLX Titanium and Illumina GaIIx next-generation sequencing platforms [8] . Whole genome shotgun sequencing and assembly provided 192,169 contigs (>500 bp) and 16,802 scaffolds (>2 kb). The N50 values, which are used to evaluate connectivity of the assembly, are 7588 bp (contigs) and 136,950 bp (scaffolds), respectively.
Cytological studies [9] [10] have reported that Thunnus species have 24 pairs of chromosomes. Draft genome sequence when merged should, therefore, only have 24 huge fragments corresponding to each chromosome. The draft genome sequence of this species is highly fragmented, and constructing a genetic linkage map is an effective way to link the sequence fragments. Genetic linkage maps have been used as an anchor for fish [11] [12] , plants [13] - [16] , and domestic animals [17] . A genetic linkage map is constructed by developing genetic markers on scaffolds and examining the linkage relationships between the markers. However, no genetic linkage map has been constructed for Pacific bluefin tuna. Several different classes of polymorphic markers could be used to construct genetic linkage maps. Repetitive sequence regions called microsatellites (MS) (or short tandem repeats) are abundant in eukaryotic genomes and have the potential to exhibit high polymorphism in a mapping population. MS markers also have the benefit of being multi-allelic within species, and therefore, unlike single nucleotide polymorphism markers (SNPs), MS markers can be used to track the unique segregation phases of both male- and female-specific parental alleles in their progeny. When parents are doubly heterozygous for SNP markers, the linkage phase of heterozygous progeny cannot be assessed. Only a few MS markers have been developed for Pacific bluefin tuna [18] , and therefore, there is a need to develop additional markers for this species.
Recently, four novel candidates for vertebrate SD genes were reported, all of them in fishes. These include amhy in the Patagonian pejerrey [19] , Gsdf in Oryzias luzonensis [20] , Amhr2 in fugu [21] and sdY in rainbow trout [22] . There are multiple sex-determining regions in teleost fishes that do not possess homology to one another due to the turnover of sex chromosomes [23] . In bluefin tuna the sex-determining region has been identified to by XY based, and male-specific marker (male delta 6 or Md6) has been characterized [24] . There is no syntenic information about Md6 and those sex-determining regions in teleost fishes. One of the objectives of this study was to localize the sex-determining locus within the genomic scaffolds corresponding to the genetic linkage map placements by mapping Md6 using an adjacent MS marker.
Comparative genome studies have revealed the evolution of chromosomes among several fish species. These studies suggest that the medaka genome has the conserved genomic structure of the MTZ ancestor (the last common ancestor of three fishes, medaka, Tetraodon, and zebrafish) and no major chromosomal rearrangements have occurred for more than 300 million years (My) [4] [25] , whereas the zebrafish genome has experienced many interchromosomal rearrangements during evolution due to extensive translocations. A comparative genome study of fugu supported their hypothesis and discussed inter-chromosomal rearrangements in the fugu and Tetraodon lineages [12] . Tongue sole also experienced three major chromosomal fusion events after divergence from the common ancestor with medaka, Tetraodon, and fugu [26] . A high density genetic map with tiled genomic platyfish contigs revealed that the platyfish and medaka karyotypes are remarkably similar with few interchromosomal translocations but with numerous intrachromosomal rearrangements (transpositions and inversions) since their lineages diverged about 120 Mya [27] . That study also suggested that stickleback and Tetraodon arose by fusion of pairs of ancestral chromosomes. These results suggest that the chromosomal constitution and synteny of fish in Percomorpha are highly conserved, with few inter-chromosomal rearrangements despite substantial phylogenetic distances among the taxa compared [28] [29] . Percomorpha is a large group in Acanthopterygii that includes medaka, platyfish, fugu, Tetraodon, stickleback, tongue sole, European seabass, and Pacific bluefin tuna. Accordingly, constructing a genetic map for other non-model fish species in Percomorpha using gene information on scaffolds from whole genome sequences will provide new aspects for comparative studies in this group.
The aim of the present study was to construct a genetic linkage map for Pacific bluefin tuna based on draft genome sequence information to understand the genome structure of this species in Percomorpha. We extracted an F1 full-sib population of young Pacific bluefin tuna as a mapping population. We constructed a linkage map using MS markers developed on each of the scaffolds and examined preservation of chromosome structure in four other fully sequenced fish species.
2. Materials and Methods
2.1. Mapping Population
Many full-sib progeny and their parents were prepared to construct a linkage map for the mapping population. Unlike other fish that spawn easily, one-to-one mating is very difficult in Pacific bluefin tuna. Thus, we tried to extract full-sib progeny from young fish spawned naturally from several parents. We collected 500 progeny (18 days old) derived from a small number of parents based on our visual observations at Amami Station, Seikai National Fisheries Research Institute, FRA. The parents of these young fish were derived from wild fish captured offshore of Shimane in the Japan Sea and reared 3 years at Amami Station, Seikai National Fisheries Research Institute, FRA. The whole bodies of the small fish were preserved in 100% ethanol until extraction of genomic DNA. DNA extraction was conducted using the Quickgene system (Fujifilm, Tokyo, Japan), following the manufacturer’s protocol. Candidate adult parents of these progeny were treated using the same method, except a fin clip was collected as the sample. Genomic DNA was amplified using the Illustra GenomiPhi Amplification Kit (GE Healthcare, Milwaukee, WI, USA) following the manufacturer’s protocol. A parental analysis of 500 young individuals was performed using 11 MS loci to select the full-sib progeny. Parent-offspring hypotheses were examined based on genotypic incompatibilities between putative parents and offspring using the exclusion method. The parentage assignment test in PARFEX software [30] was used with the exclusion method. We allowed for a few genotype mismatches (<2) between offspring and parents per MS marker due to missing data or the influence of a null allele.
2.2. Development of Microsatellite Markers
The 16,802 scaffolds that comprise the Pacific bluefin tuna genome assembly are distributed from 1.02 Mbp to 1.98 Kbp [8] . The positional information of longer scaffolds was revealed using linkage analysis during construction of the tuna physical map. Therefore, we selected the longest 1000 scaffolds as mapping candidates. Primer 3 [31] was used to design the appropriate polymerase chain reaction (PCR) primers for the near-detected MS regions that had repeat units ranging from two to five nucleotides. The size of the amplified PCR product was set to 100 - 400 bp in the reference sequence. We selected one PCR primer pairs from each scaffold, and 606 PCR primer pairs for MS were tested for polymorphism in the mapping population (Supplementary Table S1).
2.3. Genotyping and Linkage Analysis
Multiplex PCR was used to simultaneously amplify four targeted loci. The universal primer-multiplex PCR method [32] was adapted to amplify the four primer pairs. This method uses multiple universal primers each labeled with a unique fluorescent tag (FAM, VIC, NED, and PET) to co-amplify multiple loci, including size overlapping markers [32] . The sequence information for the universal primers is shown in Supplementary Table S1. We used the Type-it Microsatellite PCR kit (Qiagen, Hilden, Germany) for multiplex PCR, following the manufacturer’s recommendations. PCR amplifications were performed in a 10 µl reaction volume consisting of 5 µl Qiagen multiplex master mix, 1 µl Qiagen Q solution, 0.02 µM forward primer, 0.2 µM reverse primer, and 0.2 µM fluorescently tagged universal primers corresponding to each tailed primer. The following PCR conditions were used: initial denaturation at 94˚C for 5 min, 28 cycles of 94˚C for 30 sec, 58˚C for 90 sec and 72˚C for 30 sec, eight cycles of 94˚C for 30 sec, 53˚C for 90 sec, and 72˚C for 30 sec, followed by final extension at 59˚C for 3 min. The PCR products were heat denatured and a fragment analysis was performed on an Applied Biosystems 3130×l Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using a LIZ-600 size standard (Life Technologies, Carlsbad, CA, USA). Allele sizes were subsequently assessed and scored using GENE MAPPER ver. 4.0 (Life Technologies). After obtaining the genotyped data for each marker, the alleles were identified as paternal or maternal, which enabled construction of male-specific and female-specific linkage maps. We used MapDisto software [33] to identify each linkage group and determine the order of the MS markers in each linkage group using a log of odds threshold of 4.0. Finally, map distances were calculated using the Kosambi function. Segregation of each marker was analyzed using the chi-square test for goodness of fit to the expected Mendelian ratio in a backcross model (1:1). We focused on markers that showed significant distortion at the 5% level, after a Bonferroni correction for multiple testing. The estimated genome coverage of the map was calculated using the method 4 of Chakravarti et al. [34] , c = 1 − e−2dn/L, where d is the average interval of markers, n is the number of markers, and L is the length of the linkage map. Differences in the recombination rates between the male and female linkage maps were evaluated by calculating the interval of common contiguous markers in the female and male linkage maps.
2.4. Mapping Sex-Linked Sequences
The DNA sequence of the male characteristic fragment Md6 has been identified in cultured Pacific bluefin tuna and a BLAST search against the Pacific bluefin tuna genome assembly showed highest identity (Expect value: 8e−139) with contig BADN01109032 on scaffold Ba00007445 [24] . We designed four appropriate PCR primer sets for four MS regions on the scaffold to locate Md6 on the genetic linkage map (Supplementary Table S1). Then, we performed a linkage analysis using the same method as used for the other MS markers.
2.5. Genome Sequence and Amino Acid Sequence Comparisons
The amino acid sequence data of four teleosts (fugu, medaka, stickleback, and Tetraodon) were downloaded from the Ensembl database to construct the Oxford grid [35] . A protein BLAST search was performed for the five teleost sequences (Ensembl fishes and tuna) with an E-value < 10−5. Orthologous gene pairs were defined as one reciprocal best hit. Core genes conserved among the five teleosts were defined as the real orthologous gene set. Oxford grids were constructed to study the synteny and examine the distribution of the orthologous genes.
3. Results
3.1. Selection of the Mapping Population
Estimated parent-offspring pairs were mainly comprised of eight groups (groups 1 - 8) (Supplementary Figure S1 and Supplementary Table S2) derived from eight male and four female parents. Many individuals are needed to construct a linkage map, so we extracted 193 full-sib progeny (group 5).
3.2. Development of Microsatellite Markers
The total length of the 1000 selected scaffolds was 302 Mbp, accounting for 40.7% of all scaffolds. Of the 606 PCR primer pairs tested for MS, 473 were polymorphic in the mapping population.
3.3. Constructing the Genetic Linkage Map
Ninety-three group 5 progeny and their parents were used as the mapping population. We used 473 PCR primers to perform the linkage analysis. We constructed sex-specific maps for 24 linkage groups consisting of 470 markers (Table 1 and Figure 1). Among the informative markers used in the linkage analysis, 99.4% of the markers showed detectable linkage relationships with each other. Three markers were not linked to any other marker.

Table 1. Summary of genetic and physical map of Pacific bluefin tuna.

Figure 1. Male and female genetic linkage maps of T. orientalis. Vertical squares represents Male (left) and Female linkage group, respectively. Genetic distances between adjacent markers are shown above in Kosambi mapping function (cM). Common marker between two sexes is bridged by connected lines. Markers with red asterisks (*) showed a significant segregation distortion from the expected Mendelian 1:1 segregation in female map.
The male map consisted of 24 linkage groups with 374 MS markers. The total length of the linkage groups was 992.9 cM, and the mean distance between two markers was 2.7 cM. The sizes of individual linkage groups ranged from 5.4 to 84.2 cM (mean, 41.4 cM). The number of markers per linkage group varied from 11 to 20, with a mean of 15.6 markers per linkage group. The estimated genome coverage of the map was 86.5%.
The female map included 366 markers in 24 linkage groups. This map spanned 1332.3 cM, and mean spacing between two markers was 3.7 cM. The sizes of the female linkage groups ranged from 27.1 to 92.3 cM (mean, 55.5 cM). The number of markers per linkage group varied from 9 to 25, with a mean of 15.3 markers per group. The estimated genome coverage of the map was 86.5%.
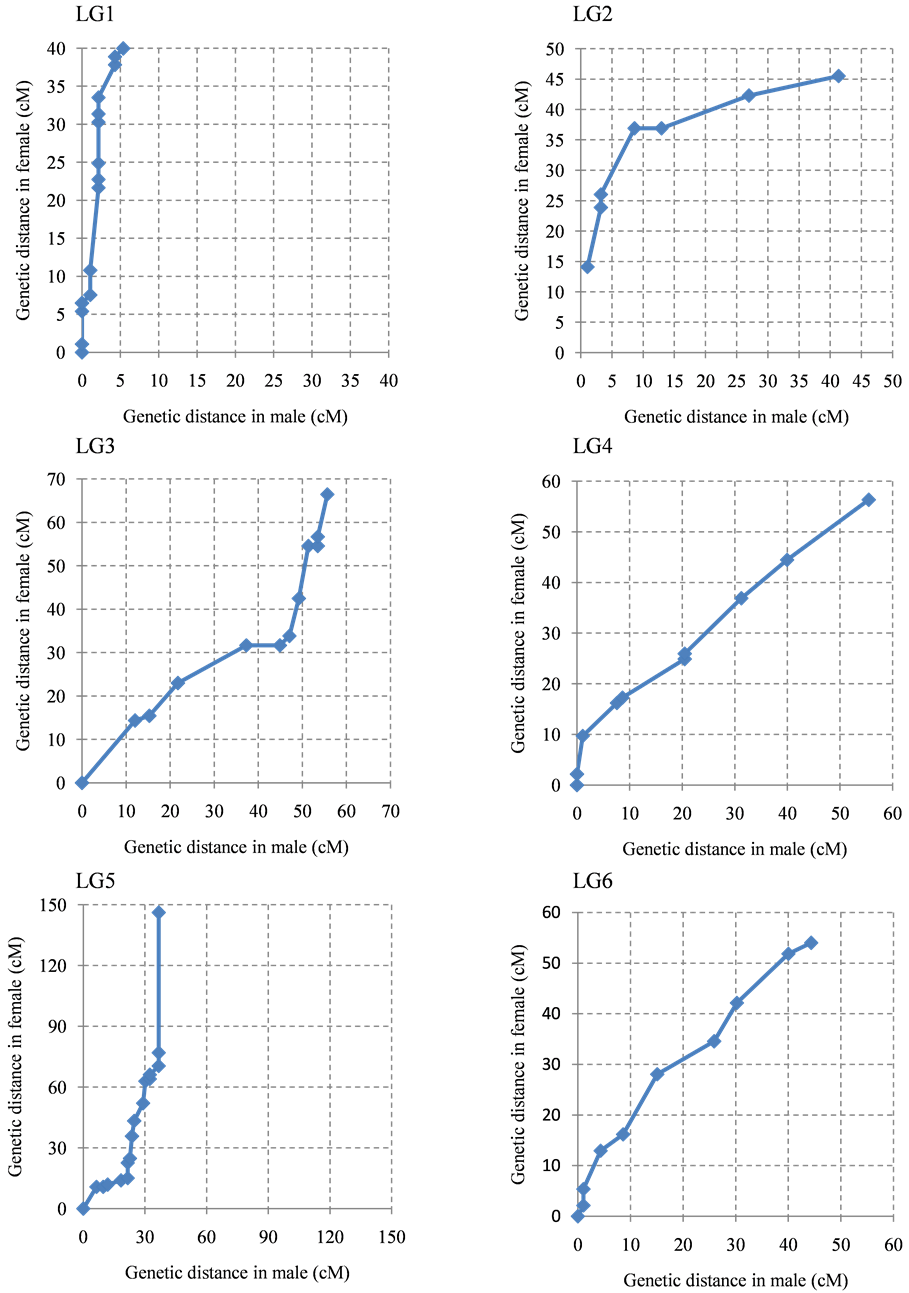
The ratio of male map length to female map length was 1.0:1.4. The distribution of the intervals between contiguous common markers in both linkage maps was almost the same (Figure 2), although several linkage groups clearly showed different recombination rates in specific regions or linkage groups (Supplementary Figure S2). In particular, the genetic distance between the common markers in LG1, LG5, LG7, LG11, LG14, LG18, and LG24 on the female map was higher than the male genetic distance. In contrast, LG15 and LG22 showed higher genetic distances on the male map.
3.4. Mapping the Sex-Linked Sequence
The MS marker (07445_1679), which was developed in scaffold Ba00007445, was only polymorphic in females and mapped to the LG10 terminal region on the female map (Figure 1). Orthologues of several sex-determining genes in teleosts [23] but were not found on the LG10 scaffold of the integrated genetic linkage map.
3.5. Integration of the Genetic Linkage Maps with the Physical Map
Developing MS markers to specific regions of each scaffold allowed us to integrate the genetic and physical maps into a consolidated genome map. We anchored 470 scaffolds to be consistent with the order of markers determined on the genetic linkage maps. Thus, we placed scaffolds that cumulatively represented 20.8% (153.8 Mb) of the sequenced genome onto the linkage groups (Table 1). In total, 4243 genes estimated in the tuna draft genome assembly have been included in these scaffolds [8] .
3.6. Comparison of Genome Structure in Other Teleosts
Constructing the integrated Pacific bluefin tuna map made it possible to compare conserved sequence regions with those of other teleosts. We identified 6445 pairs of orthologous genes among tuna and four other teleosts (fugu, medaka, stickleback, and Tetraodon). Then, we extracted 1381 gene pairs from these orthologous genes

Figure 2. Recombination ratio between male map and female map. The common intervals flanked by adjacent markers from the 24 linkage groups are compared.
and mapped them on the integrated map. We constructed an Oxford grid [35] for Pacific bluefin tuna against the four teleosts based on the number of orthologous genes on each linkage group/chromosome. These results indicated that 24 pairs of chromosomes in tuna and medaka showed a clear one-to-one relationship (Figure 3). Comparisons with the other three fish species also indicated highly conserved one-to-one relationships, although there were several one-to-two and one-to-three relationships between homologous chromosomes. For example, fugu chromosome 1 corresponded to LG3, LG19, and LG21 in Pacific bluefin tuna (Figure 3(b)). Stickleback chromosome 1 corresponded to LG2 and LG13 in Pacific bluefin tuna. A similar result was observed in stickleback chromosome 4 with LG10 and LG23 in Pacific bluefin tuna (Figure 3(c)). Three one-to-two relationships were detected between Tetraodon (chromosomes 1 - 3) and Pacific bluefin tuna (LG4 and LG10, LG19 and LG21, and LG2 and LG8) (Figure 3(d)).
We constructed an integrated map to compare the Pacific bluefin tuna and medaka chromosome structures. Homologous sequence regions across several chromosome pairs between tuna and medaka revealed reciprocal homology relationships as well as reverse orientation orderings. Thus, several inversions may have occurred in either genome (Figure 4 and Supplementary Figure S3).

 (a) (b)
(a) (b)
 (c) (d)
(c) (d)
Figure 3. Oxford grid comparing genomes of T. orientalis and four model fishes. Commonly conserved 1378 orthologous genes among five fishes were plotted into Oxford grids. Each number in a cell indicates the number of orthologs in each genome. Each grids were drawn by specific color according to the number of orthologous, The number of each linkage group of T. orientalis was determined in this study. (a) T. orientalis-Medaka comparison; (b) T. orientalis-Fugu comparison; (c) T. orientalis-Stickleback comparison; (d) T. orientalis-Tetraodon comparison.

Figure 4. Comparison of chromosome structure between Pacific bluefin tuna LG 1 and medaka Chr 1. Red number indicates MS marker name, orange square indicates homologous region between tuna and medaka. Boxes with dotted lines indicate areas in which marker order is uncertain.
4. Discussion
In this study, we constructed an integrated map composed of 24 linkage groups with 470 MS markers. The integrated map will be a useful resource to develop a complete physical map, conduct comparative genomic studies in teleosts, and perform genetic studies of economically useful traits in Pacific bluefin tuna.
The karyotype composition of Pacific bluefin tuna was two pairs of metacentric, three pairs of submetacentric, two pairs of subtelocentric, and 17 pairs of acrocentric chromosomes. The numbers of linkage groups on the sex-specific maps agreed with the number of chromosome pairs suggested from microscopic observations [9] . Scaffolds integrated with linkage groups will have to be detected on chromosome samples using fluorescence in situ hybridization to reveal the relationships between linkage groups and chromosomes, as in other fish [12] [36] . It is also desirable to locate scaffolds or contigs that include unique repeats in telomeric or centromeric regions.
Coverage of the male and female genomes by the maps was 86.5% respectively; thus, the chromosomes were covered comprehensively by this linkage map. However, mean lengths of the linkage groups were somewhat shorter (male: 41.4 cM, female: 55.5 cM) compared with linkage maps of other closely related teleosts [11] [37] - [40] , suggesting the absence of a marker at the end of a chromosome and that the mapped scaffolds lack telomeric and centromeric repeats. Restriction site-associated DNA (RAD) tag sequencing [41] using a next- generation sequencer is useful to develop several thousand single nucleotide polymorphism markers close to restriction endonuclease sites. This method has been applied to construct linkage maps for several teleosts [27] [42] - [45] . The integrated map in this study provides a framework for constructing high density-linkage map using RAD tag sequencing.
Total map length between males and females was almost the same compared with that of several other teleosts [12] [38] [46] [47] , although the female map was somewhat longer than that of the male map (ratio F:M, 1.4:1). The distributions of the common marker intervals between the male and female maps were not different.
The MD6 sex-linked sequence was mapped to the LG10 terminal region on the female linkage map. Because the mapping population in this study was 18 days old, we could not distinguish phenotypic sex. Therefore, we could not perform a linkage analysis between genotypic and phenotypic sex. A mapping population that distinguishes phenotypic sex will be needed to map the sex-determining region in the future. Sex-determining genes reported in other teleosts [23] were not found on LG10 of the integrated map. Therefore, it will be necessary to map these genes on our genetic linkage map to determine the relationships between these sex-determining genes and sex in Pacific bluefin tuna.
The comparison of chromosome structure among the five fish species suggested that Pacific bluefin tuna chromosomes were very similar to those of medaka, but traces of several inter-chromosomal rearrangements were detected in the other fish. A comprehensive amino acid sequence comparison of orthologous genes among the five teleosts (including Pacific bluefin tuna) supported that Pacific bluefin tuna is phylogenetically closer to stickleback than to medaka [8] . Taken together, Pacific bluefin tuna are more phylogenetically related to stickleback than to medaka but not in chromosome structure. The medaka genome has the conserved genomic structure of the MTZ ancestor and has not experienced any major rearrangements for more than 300 My. Our results suggest that the chromosome structure of the Pacific bluefin tuna could also be the conserved structure of the MTZ-ancestor because of the high similarity between the chromosome structures of the two species. Yellowtail (Perciformes) chromosome structure is highly similar with that of the MTZ ancestor [48] . Our results support these yellowtail results at the whole genome level.
Stickleback [27] , fugu, and Tetraodon [12] have experienced chromosome fusion events two or three times since diverging from the MTZ ancestor, which presumably had 24 chromosome pairs. Stickleback chromosome 1 corresponded to LG2 and LG13 in Pacific bluefin tuna. A similar trace was observed in stickleback chromosome 4 with LG10 and LG23 in Pacific bluefin tuna. These one-to-two relationships are potential traces of inter-chromosomal rearrangements in a lineage phylogenetically diverged from the two species. In this case, fusion of two chromosomes or chromosome fission in the stickleback lineage could have occurred after divergence from the common ancestor. Our comparative study did not consider intra-chromosomal rearrangements that diverged from the MTZ ancestor. An investigation of macro or micro-synteny among fish species in which whole genome sequences are available is needed to understand the detailed characteristics of Pacific bluefin tuna chromosome structure and to estimate rearrangement events in the Perciformes lineage.
It was a surprising and new finding that medaka and Pacific bluefin tuna have similar chromosome constitutions, even though these two species are evolutionary distant in Percomorpha and differ morphologically and physiologically. This result suggests that other fish in Percomorpha may have a similar chromosome constitution and traces of inter-chromosomal changes. It is also possible that chromosome constitution does not always reflect phylogenetic distance based on other genetic factors, such as nucleotide or amino acid sequences. Several chromosome pairs and homologous sequence regions between tuna and medaka were in reverse orientation between these two species. Chromosome inversion may have occurred from ectopic recombination between homologous sequences or breakage of chromosomes and erroneous repair of free ends by non-homologous end- joining [49] . Genes at recombination breakpoints occasionally experience a fission or fusion event with other genes during chromosomal inversion, fusion, or translocation. Such a phenomenon has been explained as the mechanism of novel gene birth [49] . Thus, different genes are detected in these boundary regions of Pacific bluefin tuna and medaka.
Pacific bluefin tuna has high market value, but wild populations have decreased in recent years [1] . The complete lifecycle of this species has been closed for aquaculture, and artificial seed is being used for production [2] . Therefore, there is interest in creating brood stocks with commercially valuable traits, such as rapid growth. A genetic linkage map with many DNA markers is needed to efficiently find markers associated with quantitative trait loci (QTL) that can be used in marker-assisted selection breeding programs to genetically improve traits, such as disease resistance, high growth rate, and sex determination in Pacific bluefin tuna seed and seedlings. QTL analyses [50] for specific traits have been performed in many fish including groups phylogenetically close to Pacific bluefin tuna in Percomorpha, such as two yellowtail species (Seriola quinqueradiata and Seriola lalandi) in Carangidae [37] [51] and Japanese flounder (Paralichthys olivaceus) and Atlantic halibut (Hippoglossus hippoglossus) in Pleuronectiformes [45] [52] [53] . In this study, we developed sex-specific linkage maps using a tuna draft genome sequence. The mean distances between two markers in the male and female maps were 2.7 cM and 3.7 cM, respectively. This marker density is sufficient to perform a rough QTL analysis [54] . The linkage map will be a useful resource for QTL analyses of economically useful traits in this species. Moreover, it could be used to elucidate the genetic basis of the ecologically unique traits of Pacific bluefin tuna. Integrated sequence information from the linkage maps will help identify responsible genes in candidate regions detectable by QTL analysis.
Acknowledgements
The authors acknowledge Amami Fish Breeding (Maruha Nichiro) for providing samples during MS marker development. We thank the members of the Research Center for Tuna Aquaculture, Seikai National Fisheries Research Institute (Fisheries Research Agency) for providing the tuna mapping samples. This study was supported by the bluefin tuna breeding program at the Fisheries Agency.
Cite this paper
TsubasaUchino,YojiNakamura,MasashiSekino,WataruKai,AtushiFujiwara,MotoshigeYasuike,TakumaSugaya,HimekoFukuda,MotohikoSano,TakashiSakamoto, (2016) Constructing Genetic Linkage Maps Using the Whole Genome Sequence of Pacific Bluefin Tuna (Thunnus orientalis) and a Comparison of Chromosome Structure among Teleost Species. Advances in Bioscience and Biotechnology,07,85-122. doi: 10.4236/abb.2016.72010
References
- 1. Masuma, S., Takebe, T. and Sakakura, Y. (2011) A Review of the Broodstock Management and Larviculture of the Pacific Northern Bluefin Tuna in Japan. Aquaculture, 315, 2-8. http://dx.doi.org/10.1016/j.aquaculture.2010.05.030
- 2. Sawada, Y., Okada, T., Miyashita, S., Murata, O. and Kumai, H. (2005) Completion of the Pacific Bluefin Tuna Thunnus orientalis (Temminck et Schlegel) Life Cycle. Aquaculture Research, 36, 413-421.
http://dx.doi.org/10.1111/j.1365-2109.2005.01222.x - 3. Howe, K., Clark, M.D., Torroja, C.F., Torrance, J., Berthelot, C., Muffato, M., Collins, J.E.J., Humphray, S., McLaren, K., Matthews, L., McLaren, S., Sealy, I., Caccamo, M., Churcher, C., Scott, C., Barrett, J.C., Koch, R., Rauch, G.-J., White, S., Chow, W., Kilian, B., Quintais, L.T., Guerra-Assunção, J.A., Zhou, Y., Gu, Y., Yen, J., Vogel, J.-H., Eyre, T., Redmond, S., Banerjee, R., Chi, J., Fu, B., Langley, E., Maguire, S.F., Laird, G.K., Lloyd, D., Kenyon, E., Donaldson, S., Sehra, H., Almeida-King, J., Loveland, J., Trevanion, S., Jones, M., Quail, M., Willey, D., Hunt, A., Burton, J., Sims, S., McLay, K., Plumb, B., Davis, J., Clee, C., Oliver, K., Clark, R., Riddle, C., Elliot, D., Eliott, D., Threadgold, G., Harden, G., Ware, D., Mortimore, B., Mortimer, B., Kerry, G., Heath, P., Phillimore, B., Tracey, A., Corby, N., Dunn, M., Johnson, C., Wood, J., Clark, S., Pelan, S., Griffiths, G., Smith, M., Glithero, R., Howden, P., Barker, N., Stevens, C., Harley, J., Holt, K., Panagiotidis, G., Lovell, J., Beasley, H., Henderson, C., Gordon, D., Auger, K., Wright, D., Raisen, C., Dyer, L., Leung, K., Robertson, L., Ambridge, K., Leongamornlert, D., McGuire, S., Gilderthorp, R., Griffiths, C., Manthravadi, D., Nichol, S., Barker, G., Whitehead, S., Kay, M., Brown, J., Murnane, C., Gray, E., Humphries, M., Sycamore, N., Barker, D., Saunders, D., Wallis, J., Babbage, A., Hammond, S., Mashreghi-Mohammadi, M., Barr, L., Martin, S., Wray, P., Ellington, A., Matthews, N., Ellwood, M., Woodmansey, R., Clark, G., Cooper, J.D., Tromans, A., Grafham, D., Skuce, C., Pandian, R., Andrews, R., Harrison, E., Kimberley, A., Garnett, J., Fosker, N., Hall, R., Garner, P., Kelly, D., Bird, C., Palmer, S., Gehring, I., Berger, A., Dooley, C.M., Ersan-ürün, Z., Eser, C., Geiger, H., Geisler, M., Karotki, L., Kirn, A., Konantz, J., Konantz, M., Oberländer, M., Rudolph-Geiger, S., Teucke, M., Osoegawa, K., Zhu, B., Rapp, A., Widaa, S., Langford, C., Yang, F., Carter, N.P., Harrow, J., Ning, Z., Herrero, J., Searle, S.M.J., Enright, A., Geisler, R., Plasterk, R.H.A., Lee, C., Westerfield, M., de Jong, P.J., Zon, L.I., Postlethwait, J.H., Nüsslein-Volhard, C., Hubbard, T.J.P., Roest Crollius, H., Rogers, J., Stemple, D.L., Begum, S., Lloyd, C., Lanz, C., Raddatz, G. and Schuster, S.C. (2013) The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature, 496, 498-503.
- 4. Kasahara, M., Naruse, K., Sasaki, S., Nakatani, Y., Qu, W., Ahsan, B., Yamada, T., Nagayasu, Y., Doi, K., Kasai, Y., Jindo, T., Kobayashi, D., Shimada, A., Toyoda, A., Kuroki, Y., Fujiyama, A., Sasaki, T., Shimizu, A., Asakawa, S., Shimizu, N., Hashimoto, S.-I., Yang, J., Lee, Y., Matsushima, K., Sugano, S., Sakaizumi, M., Narita, T., Ohishi, K., Haga, S., Ohta, F., Nomoto, H., Nogata, K., Morishita, T., Endo, T., Shin-I, T., Takeda, H., Morishita, S. and Kohara, Y. (2007) The Medaka Draft Genome and Insights into Vertebrate Genome Evolution. Nature, 447, 714-719.
http://dx.doi.org/10.1038/nature05846 - 5. Aparicio, S., Chapman, J., Stupka, E., Putnam, N., Chia, J.-M., Dehal, P., Christoffels, A., Rash, S., Hoon, S., Smit, A., Gelpke, M.D.S., Roach, J., Oh, T., Ho, I.Y., Wong, M., Detter, C., Verhoef, F., Predki, P., Tay, A., Lucas, S., Richardson, P., Smith, S.F., Clark, M.S., Edwards, Y.J.K., Doggett, N., Zharkikh, A., Tavtigian, S.V., Pruss, D., Barnstead, M., Evans, C., Baden, H., Powell, J., Glusman, G., Rowen, L., Hood, L., Tan, Y.H., Elgar, G., Hawkins, T., Venkatesh, B., Rokhsar, D. and Brenner, S. (2002) Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu rubripes. Science (New York, N.Y.), 297, 1301-1310.
- 6. Jaillon, O., Aury, J.-M., Brunet, F., Petit, J.-L., Stange-Thomann, N., Mauceli, E., Bouneau, L., Fischer, C., Ozouf-Costaz, C., Bernot, A., Nicaud, S., Jaffe, D., Fisher, S., Lutfalla, G., Dossat, C., Segurens, B., Dasilva, C., Salanoubat, M., Levy, M., Boudet, N., Castellano, S., Anthouard, V., Jubin, C., Castelli, V., Katinka, M., Vacherie, B., Biémont, C., Skalli, Z., Cattolico, L., Poulain, J., De Berardinis, V., Cruaud, C., Duprat, S., Brottier, P., Coutanceau, J.-P., Gouzy, J., Parra, G., Lardier, G., Chapple, C., McKernan, K.J., McEwan, P., Bosak, S., Kellis, M., Volff, J.-N., Guigó, R., Zody, M. C., Mesirov, J., Lindblad-Toh, K., Birren, B., Nusbaum, C., Kahn, D., Robinson-Rechavi, M., Laudet, V., Schachter, V., Quétier, F., Saurin, W., Scarpelli, C., Wincker, P., Lander, E.S., Weissenbach, J. and Roest Crollius, H. (2004) Genome Duplication in the Teleost Fish Tetraodon nigroviridis Reveals the Early Vertebrate Proto-Karyotype. Nature, 431, 946-957.
http://dx.doi.org/10.1038/nature03025 - 7. Jones, F.C., Grabherr, M.G., Chan, Y.F., Russell, P., Mauceli, E., Johnson, J., Swofford, R., Pirun, M., Zody, M.C., White, S., Birney, E., Searle, S., Schmutz, J., Grimwood, J., Dickson, M.C., Myers, R.M., Miller, C.T., Summers, B.R., Knecht, A.K., Brady, S.D., Zhang, H., Pollen, A.A., Howes, T., Amemiya, C., Baldwin, J., Bloom, T., Jaffe, D.B., Nicol, R., Wilkinson, J., Lander, E.S., Di Palma, F., Lindblad-Toh, K. and Kingsley, D.M. (2012) The Genomic Basis of Adaptive Evolution in Three Spine Sticklebacks. Nature, 484, 55-61.
http://dx.doi.org/10.1038/nature10944 - 8. Nakamura, Y., Mori, K., Saitoh, K., Oshima, K., Mekuchi, M., Sugaya, T., Shigenobu, Y., Ojima, N., Muta, S., Fujiwara, A., Yasuike, M., Oohara, I., Hirakawa, H., Chowdhury, V.S., Kobayashi, T., Nakajima, K., Sano, M., Wada, T., Tashiro, K., Ikeo, K., Hattori, M., Kuhara, S., Gojobori, T. and Inouye, K. (2013) Evolutionary Changes of Multiple Visual Pigment Genes in the Complete Genome of Pacific Bluefin Tuna. Proceedings of the National Academy of Sciences of the United States of America, 110, 11061-11066.
http://dx.doi.org/10.1073/pnas.1302051110 - 9. Ida, H., Oka, N. and Hayashigaki, K. (1991) Karyotypes and Cellular DNA Contents of Three Species of the Subfam ily Clupeinae. Japanese Journal of Ichthyology, 38, 289-294.
- 10. Soares, R.X., Bertollo, L.A.C., Costa, G.W.W.F. and Molina, W.F. (2013) Karyotype Stasis in Four Atlantic Scombridae Fishes: Mapping of Classic and Dual-Color FISH Markers on Chromosomes. Fisheries Science, 79, 177-183.
http://dx.doi.org/10.1007/s12562-013-0602-0 - 11. Kai, W., Kikuchi, K., Fujita, M., Suetake, H., Fujiwara, A., Yoshiura, Y., Ototake, M., Venkatesh, B., Miyaki, K. and Suzuki, Y. (2005) A Genetic Linkage Map for the Tiger Puffer Fish, Takifugu rubripes. Genetics, 171, 227-238.
http://dx.doi.org/10.1534/genetics.105.042051 - 12. Kai, W., Kikuchi, K., Tohari, S., Chew, A.K., Tay, A., Fujiwara, A., Hosoya, S., Suetake, H., Naruse, K., Brenner, S., Suzuki, Y. and Venkatesh, B. (2011) Integration of the Genetic Map and Genome Assembly of fugu Facilitates Insights into Distinct Features of Genome Evolution in Teleosts and Mammals. Genome Biology and Evolution, 3, 424-442.
http://dx.doi.org/10.1093/gbe/evr041 - 13. Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., Kato, T., Nakao, M., Sasamoto, S., Watanabe, A., Ono, A., Ka washima, K., Fujishiro, T., Katoh, M., Kohara, M., Kishida, Y., Minami, C., Nakayama, S., Nakazaki, N., Shimizu, Y., Shinpo, S., Takahashi, C., Wada, T., Yamada, M., Ohmido, N., Hayashi, M., Fukui, K., Baba, T., Nakamichi, T., Mori, H. and Tabata, S. (2008) Genome Structure of the Legume, Lotus japonicas. DNA Research, 15, 227-239.
http://dx.doi.org/10.1093/dnares/dsn008 - 14. Ren, Y., Zhao, H., Kou, Q., Jiang, J., Guo, S., Zhang, H., Hou, W., Zou, X., Sun, H., Gong, G., Levi, A. and Xu, Y. (2012) A High Resolution Genetic Map Anchoring Scaffolds of the Sequenced Watermelon Genome. PloS One, 7, e29453.
http://dx.doi.org/10.1371/journal.pone.0029453 - 15. Yang, H., Tao, Y., Zheng, Z., Zhang, Q., Zhou, G., Sweetingham, M.W., Howieson, J.G. and Li, C. (2013) Draft Genome Sequence, and a Sequence-Defined Genetic Linkage Map of the Legume Crop Species Lupinus angustifolius L. PloS One, 8, e64799.
- 16. Ma, X.-F., Jensen, E., Alexandrov, N., Troukhan, M., Zhang, L., Thomas-Jones, S., Farrar, K., Clifton-Brown, J., Donnison, I., Swaller, T. and Flavell, R. (2012) High Resolution Genetic Mapping by Genome Sequencing Reveals Genome Duplication and Tetraploid Genetic Structure of the Diploid Miscanthus sinensis. PloS One, 7, e33821.
http://dx.doi.org/10.1371/journal.pone.0033821 - 17. Wong, A.K., Ruhe, A.L., Dumont, B.L., Robertson, K.R., Guerrero, G., Shull, S.M., Ziegle, J.S., Millon, L.V., Broman, K.W., Payseur, B.A. and Neff, M.W. (2010) A Comprehensive Linkage Map of the Dog Genome. Genetics, 184, 595-605.
http://dx.doi.org/10.1534/genetics.109.106831 - 18. Morishima, K., Yamamoto, H., Sawada, Y., Miyashita, S. and Kato, K. (2009) Developing 23 New Polymorphic Microsatellite Markers and Simulating Parentage Assignment in the Pacific Bluefin Tuna, Thunnus orientalis. Molecular Ecology Resources, 9, 790-792.
http://dx.doi.org/10.1111/j.1755-0998.2008.02144.x - 19. Hattori, R.S., Murai, Y., Oura, M., Masuda, S., Majhi, S.K., Sakamoto, T., Fernandino, J.I., Somoza, G.M., Yokota, M. and Strussmann, C.A. (2012) A Y-Linked Anti-Mullerian Hormone Duplication Takes over a Critical Role in Sex Determination. Proceedings of the National Academy of Sciences, 109, 2955-2959.
http://dx.doi.org/10.1073/pnas.1018392109 - 20. Myosho, T., Otake, H., Masuyama, H., Matsuda, M., Kuroki, Y., Fujiyama, A., Naruse, K., Hamaguchi, S. and Sakaizumi, M. (2012) Tracing the Emergence of a Novel Sex-Determining Gene in Medaka, Oryzias luzonensis. Genetics, 191, 163-170.
http://dx.doi.org/10.1534/genetics.111.137497 - 21. Kamiya, T., Kai, W., Tasumi, S., Oka, A., Matsunaga, T., Mizuno, N., Fujita, M., Suetake, H., Suzuki, S., Hosoya, S., Tohari, S., Brenner, S., Miyadai, T., Venkatesh, B., Suzuki, Y. and Kikuchi, K. (2012) A Trans-Species Missense SNP in Amhr2 Is Associated with Sex Determination in the Tiger Pufferfish, Takifugu rubripes (Fugu). PLoS Genetics, 8, e1002798.
http://dx.doi.org/10.1371/journal.pgen.1002798 - 22. Yano, A., Nicol, B., Jouanno, E., Quillet, E., Fostier, A., Guyomard, R. and Guiguen, Y. (2013) The Sexually Dimorphic on the Y-Chromosome Gene (sdY) Is a Conserved Male-Specific Y-Chromosome Sequence in Many Salmonids. Evolutionary Applications, 6, 486-496.
http://dx.doi.org/10.1111/eva.12032 - 23. Kikuchi, K. and Hamaguchi, S. (2013) Novel Sex-Determining Genes in Fish and Sex Chromosome Evolution. Developmental Dynamics, 242, 339-353.
http://dx.doi.org/10.1002/dvdy.23927 - 24. Agawa, Y., Iwaki, M., Komiya, T., Honryo, T., Tamura, K., Okada, T., Yagishita, N., Kobayashi, T. and Sawada, Y. (2014) Identification of Male Sex-Linked DNA Sequence of the Cultured Pacific Bluefin Tuna Thunnus orientalis. Fisheries Science, 81, 113-121.
http://dx.doi.org/10.1007/s12562-014-0833-8 - 25. Naruse, K., Tanaka, M., Mita, K., Shima, A., Postlethwait, J. and Mitani, H. (2004) A Medaka Gene Map: The Trace of Ancestral Vertebrate Proto-Chromosomes Revealed by Comparative Gene Mapping. Genome Research, 14, 820-828.
http://dx.doi.org/10.1101/gr.2004004 - 26. Chen, S., Zhang, G., Shao, C., Huang, Q., Liu, G., Zhang, P., Song, W., An, N., Chalopin, D., Volff, J.-N., Hong, Y., Li, Q., Sha, Z., Zhou, H., Xie, M., Yu, Q., Liu, Y., Xiang, H., Wang, N., Wu, K., Yang, C., Zhou, Q., Liao, X., Yang, L., Hu, Q., Zhang, J., Meng, L., Jin, L., Tian, Y., Lian, J., Yang, J., Miao, G., Liu, S., Liang, Z., Yan, F., Li, Y., Sun, B., Zhang, H., Zhang, J., Zhu, Y., Du, M., Zhao, Y., Schartl, M., Tang, Q. and Wang, J. (2014) Whole-Genome Sequence of A Flatfish Provides Insights into ZW Sex Chromosome Evolution and Adaptation to a Benthic Lifestyle. Nature Genetics, 46, 253-260.
http://dx.doi.org/10.1038/ng.2890 - 27. Amores, A., Catchen, J., Nanda, I., Warren, W., Walter, R., Schartl, M. and Postlethwait, J.H. (2014) A RAD-Tag Genetic Map for the Platyfish (Xiphophorus maculatus) Reveals Mechanisms of Karyotype Evolution among Teleost Fish. Genetics, 197, 625-641.
http://dx.doi.org/10.1534/genetics.114.164293 - 28. Kawahara, R., Miya, M., Mabuchi, K., Lavoué, S., Inoue, J.G., Satoh, T.P., Kawaguchi, A. and Nishida, M. (2008) Interrelationships of the 11 Gasterosteiform Families (Sticklebacks, Pipefishes, and Their Relatives): A New Perspective Based on Whole Mitogenome Sequences from 75 Higher Teleosts. Molecular Phylogenetics and Evolution, 46, 224-236.
http://dx.doi.org/10.1016/j.ympev.2007.07.009 - 29. Setiamarga, D.H.E., Miya, M., Yamanoue, Y., Mabuchi, K., Satoh, T.P., Inoue, J.G. and Nishida, M. (2008) In Terrelationships of Atherinomorpha (Medakas, Flyingfishes, Killifishes, Silversides, and Their Relatives): The First Evidence Based on Whole Mitogenome Sequences. Molecular Phylogenetics and Evolution, 49, 598-605.
http://dx.doi.org/10.1016/j.ympev.2008.08.008 - 30. Sekino, M. and Kakehi, S. (2011) PARFEX v1.0: An EXCELTM-Based Software Package for Parentage Allocation. Conservation Genetics Resources, 4, 275-278.
http://dx.doi.org/10.1007/s12686-011-9523-3 - 31. Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B.C., Remm, M. and Rozen, S.G. (2012) Primer3— New Capabilities and Interfaces. Nucleic Acids Research, 40, e115.
http://dx.doi.org/10.1093/nar/gks596 - 32. Blacket, M.J., Robin, C., Good, R.T., Lee, S.F. and Miller, A.D. (2012) Universal Primers for Fluorescent Label Ling of PCR Fragments—An Efficient and Cost-Effective Approach to Genotyping by Fluorescence. Molecular Ecology Resources, 12, 456-463.
http://dx.doi.org/10.1111/j.1755-0998.2011.03104.x - 33. Lorieux, M. (2012) MapDisto: Fast and Efficient Computation of Genetic Linkage Maps. Molecular Breeding, 30, 1231-1235.
http://dx.doi.org/10.1007/s11032-012-9706-y - 34. Chakravarti, A., Lasher, L.K. and Reefer, J.E. (1991) A Maximum Likelihood Method for Estimating Genome Length Using Genetic Linkage Data. Genetics, 128, 175-182.
- 35. Edwards, J.H. (1991) The Oxford Grid. Annals of Human Genetics, 55, 17-31.
http://dx.doi.org/10.1111/j.1469-1809.1991.tb00394.x - 36. Fujiwara, A., Fujiwara, M., Nishida-Umehara, C., Abe, S. and Masaoka, T. (2007) Characterization of Japanese Flounder Karyotype by Chromosome Bandings and Fluorescence in Situ Hybridization with DNA Markers. Genetica, 131, 267-274.
http://dx.doi.org/10.1007/s10709-006-9136-z - 37. Ohara, E., Nishimura, T., Nagakura, Y., Sakamoto, T., Mushiake, K. and Okamoto, N. (2005) Genetic Linkage Maps of Two Yellowtails (Seriola quinqueradiata and Seriola lalandi). Aquaculture, 244, 41-48.
http://dx.doi.org/10.1016/j.aquaculture.2004.10.022 - 38. Reid, D.P., Smith, C.-A., Rommens, M., Blanchard, B., Martin-Robichaud, D. and Reith, M. (2007) A Genetic Linkage Map of Atlantic Halibut (Hippoglossus hippoglossus L.). Genetics, 177, 1193-1205.
http://dx.doi.org/10.1016/j.aquaculture.2004.10.022 - 39. Castaño-Sánchez, C., Fuji, K., Ozaki, A., Hasegawa, O., Sakamoto, T., Morishima, K., Nakayama, I., Fujiwara, A., Masaoka, T., Okamoto, H., Hayashida, K., Tagami, M., Kawai, J., Hayashizaki, Y. and Okamoto, N. (2010) A Second Generation Genetic Linkage Map of Japanese Flounder (Paralichthys olivaceus). BMC Genomics, 11, 554.
http://dx.doi.org/10.1186/1471-2164-11-554 - 40. Fuji, K., Koyama, T., Kai, W., Kubota, S., Yoshida, K., Ozaki, A., Aoki, J., Kawabata, Y., Araki, K., Tsuzaki, T., Okamoto, N. and Sakamoto, T. (2014) Construction of a High-Coverage Bacterial Artificial Chromosome Library and Comprehensive Genetic Linkage Map of Yellowtail Seriola quinqueradiata. BMC Research Notes, 7, 200.
http://dx.doi.org/10.1186/1756-0500-7-200 - 41. Baird, N.A., Etter, P.D., Atwood, T.S., Currey, M.C., Shiver, A.L., Lewis, Z.A., Selker, E.U., Cresko, W.A. and Johnson, E.A. (2008) Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PloS One, 3, e3376.
http://dx.doi.org/10.1371/journal.pone.0003376 - 42. Amores, A., Catchen, J., Ferrara, A., Fontenot, Q. and Postlethwait, J.H. (2011) Genome Evolution and Meiotic Maps by Massively Parallel DNA Sequencing: Spotted Gar, an Outgroup for the Teleost Genome Duplication. Genetics, 188, 799-808.
http://dx.doi.org/10.1534/genetics.111.127324 - 43. Everett, M.V., Miller, M.R. and Seeb, J.E. (2012) Meiotic Maps of Sockeye Salmon Derived from Massively Parallel DNA Sequencing. BMC Genomics, 13, 521.
http://dx.doi.org/10.1186/1471-2164-13-521 - 44. Kakioka, R., Kokita, T., Kumada, H., Watanabe, K. and Okuda, N. (2013) A RAD-Based Linkage Map and Comparative Genomics in the Gudgeons (Genus Gnathopogon, Cyprinidae). BMC Genomics, 14, 32.
http://dx.doi.org/10.1186/1471-2164-14-32 - 45. Palaiokostas, C., Bekaert, M., Davie, A., Cowan, M.E., Oral, M., Taggart, J.B., Gharbi, K., McAndrew, B.J., Penman, D.J. and Migaud, H. (2013) Mapping the Sex Determination Locus in the Atlantic Halibut (Hippoglossus hippoglossus) Using RAD Sequencing. BMC Genomics, 14, 566.
http://dx.doi.org/10.1186/1471-2164-14-566 - 46. Sakamoto, T., Danzmann, R.G., Gharbi, K., Howard, P., Ozaki, A., Khoo, S.K., Woram, R.A., Okamoto, N., Ferguson, M.M., Holm, L.E., Guyomard, R. and Hoyheim, B. (2000) A Microsatellite Linkage Map of Rainbow Trout (Oncorhynchus mykiss) Characterized by Large Sex-Specific Differences in Recombination Rates. Genetics, 155, 1331-1345.
- 47. Singer, A., Perlman, H., Yan, Y., Walker, C., Corley-Smith, G., Brandhorst, B. and Postlethwait, J. (2002) Sex-Specific Recombination Rates in Zebrafish (Danio rerio). Genetics, 160, 649-657.
- 48. Aoki, J.-Y., Kai, W., Kawabata, Y., Ozaki, A., Yoshida, K., Koyama, T., Sakamoto, T. and Araki, K. (2015) Second Generation Physical and Linkage Maps of Yellowtail (Seriola quinqueradiata) and Comparison of Synteny with Four Model Fish. BMC Genomics, 16, 406. http://dx.doi.org/10.1186/s12864-015-1600-7
- 49. Guillén, Y. and Ruiz, A. (2012) Gene Alterations at Drosophila Inversion Breakpoints Provide Prima Facie Evidence for Natural Selection as an Explanation for Rapid Chromosomal Evolution. BMC Genomics, 13, 53.
http://dx.doi.org/10.1186/1471-2164-13-53 - 50. Yue, G.H. (2013) Recent Advances of Genome Mapping and Marker-Assisted Selection in Aquaculture. Fish and Fisheries, 15, 376-396.
http://dx.doi.org/10.1111/faf.12020 - 51. Ozaki, A., Yoshida, K., Fuji, K., Kubota, S., Kai, W., Aoki, J., Kawabata, Y., Suzuki, J., Akita, K., Koyama, T., Nakagawa, M., Hotta, T., Tsuzaki, T., Okamoto, N., Araki, K. and Sakamoto, T. (2013) Quantitative Trait Loci (QTL) Associated with Resistance to a Monogenean Parasite (Benedenia seriolae) in Yellowtail (Seriola quinqueradiata) through Genome Wide Analysis. PloS One, 8, e64987.
http://dx.doi.org/10.1371/journal.pone.0064987 - 52. Fuji, K., Kobayashi, K., Hasegawa, O., Coimbra, M.R.M., Sakamoto, T. and Okamoto, N. (2006) Identification of a Single Major Genetic Locus Controlling the Resistance to Lymphocystis Disease in Japanese Flounder (Paralichthys olivaceus). Aquaculture, 254, 203-210.
http://dx.doi.org/10.1016/j.aquaculture.2005.11.024 - 53. Song, W., Pang, R., Niu, Y., Gao, F., Zhao, Y., Zhang, J., Sun, J., Shao, C., Liao, X., Wang, L., Tian, Y. and Chen, S. (2012) Construction of High-Density Genetic Linkage Maps and Mapping of Growth-Related Quantitative Trail Loci in the Japanese Flounder (Paralichthys olivaceus). PloS One, 7, e50404.
http://dx.doi.org/10.1371/journal.pone.0050404 - 54. Broman, K.W. (1999) Review of Statistical Methods for QTL Mapping in Experimental Crosses. Lab Animal, 30, 44-52.
Supplementary Figures and Tables
Table S1. The information of PCR primers used in this study. These primers are categorized as follows. A: used in parentage test. B: used in linkage mapping.
Table S2. Result of parentage test.

Figure S1. Composition of the full-sib population estimated from about 500 Pacific bluefin tuna progeny.
 (a)
(a)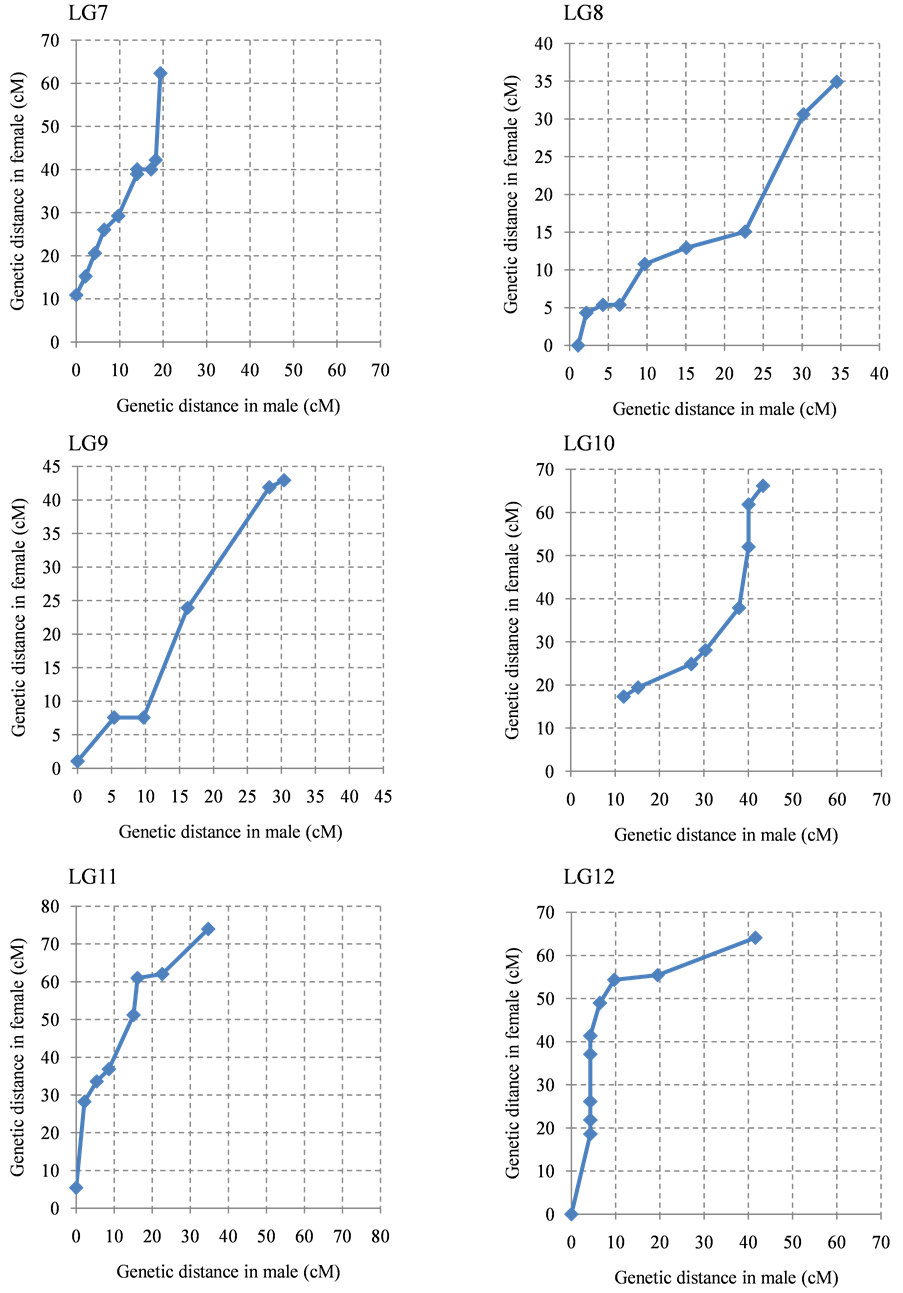 (b)
(b)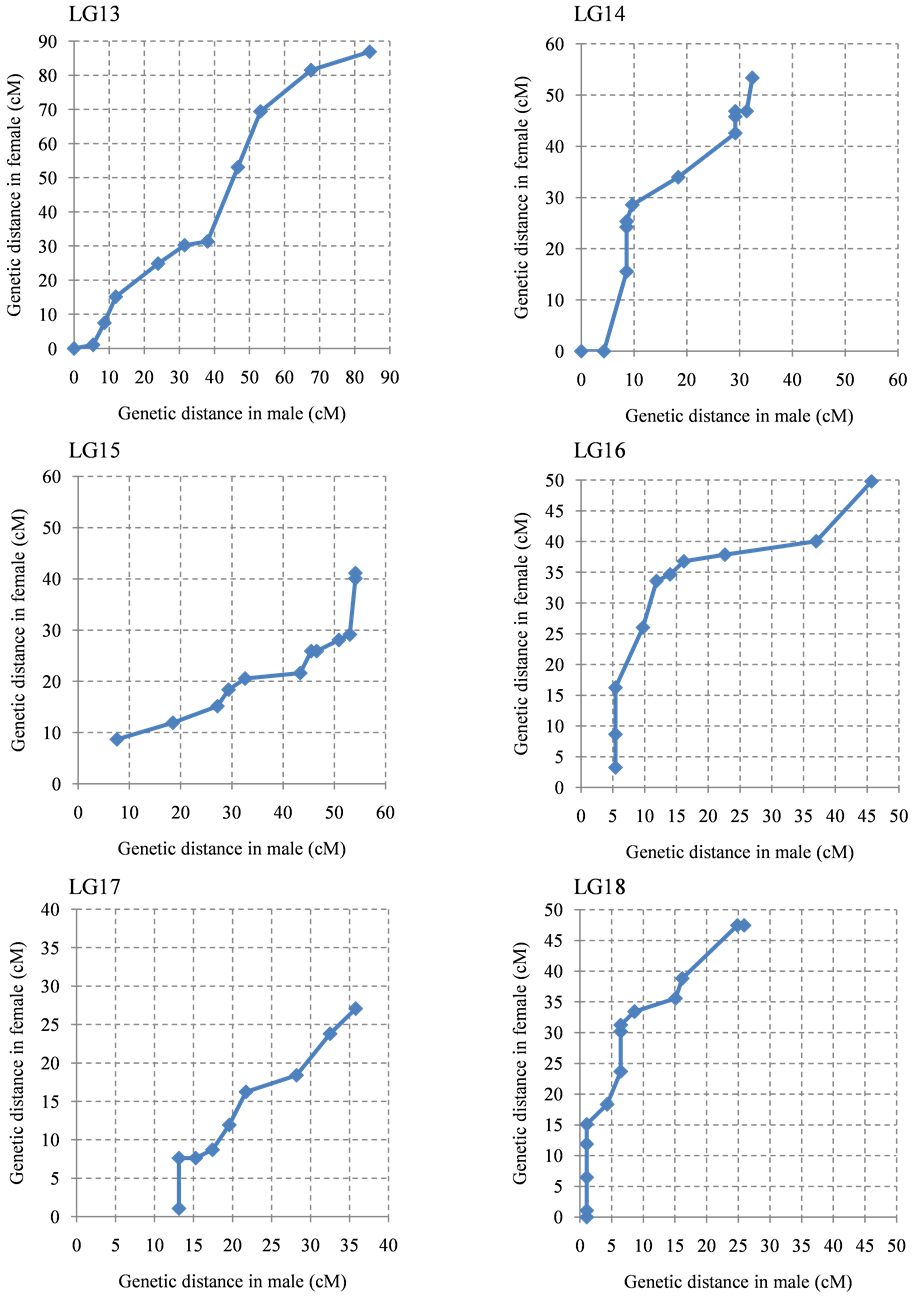 (c)
(c) (d)
(d)
Figure S2. The intervals of between contiguous common marker in male linkage map and female linkage map. Contiguous common markers between both linkage group were plotted by dot. X axis and Y axis show cumulative genetic distance of linkage group of male and female, respectively. (a) LG 1-LG6; (b) LG7-LG12; (c) LG13-LG18; (d) LG19-LG24.

Figure S3. Comparison of chromosome structure between bluefin tuna and medaka. Red number indicates MS marker name, orange square indicates homologous region between tuna and medaka. Boxes with dotted lines indicate areas in which marker order is uncertain.
NOTES

*Corresponding author.
上一篇:Online Effective Identificatio 下一篇:Mitochondria: transportation,
最新文章NEWS
- Research on the Repeated Sequences among tRNA Sequences
- MicroRNA-21, 204 and 125b Play Potential Roles in Tumorigenesis of Melanoma
- Agrobacterium-Mediated Transformation of Mexican Lime (Citrus aurantifolia Swingle) Using Optimized
- Quality Control of Selected Antimalarials Sold in the Illicit Market: An Investigation Conducted in
- Structure of Ulvan Isolated from the Edible Green Seaweed, Ulva pertusa
- Effects of Media Composition and Auxins on Adventitious Rooting of Bienertia sinuspersici Cuttings
- Online Effective Identification of Glycopeptide Using Liquid Chromatography Combined with Fourier Tr
- Behavior and Viability of Blueberry Seeds through Germination and Tetrazolium Test
推荐期刊Tui Jian
- Chinese Journal of Integrative Medicine
- Journal of Genetics and Genomics
- Journal of Bionic Engineering
- Pedosphere
- Chinese Journal of Structural Chemistry
- Nuclear Science and Techniques
- 《传媒》
- 《中学生报》教研周刊
热点文章HOT
- Metal Salts Assisted Enzyme-Based Extraction of Stevioside from the Leaves of Stevia rebaudiana Bert
- Efficient Production of δ-Guaiene, an Aroma Sesquiterpene Compound Accumulated in Agarwood, by Meval
- Agrobacterium-Mediated Transformation of Mexican Lime (Citrus aurantifolia Swingle) Using Optimized
- Structure of Alpha-Gliadin Multigene and Construction of Efficient Hairpin RNAi Molecule against Glu
- cDNA Cloning of Paramyosin from Several Kinds of Squid Mantle Muscle
- Biotechnological Transformation of Lignocellulosic Biomass in to Industrial Products: An Overview
- Transforming the Snapdragon Aurone Biosynthetic Genes into Petunia Alters Coloration Patterns in Tra
- Role of Probiotics in Pancreatic Cancer Prevention: The Prospects and Challenges