
A Risk Coefficient for Radiation-Induced Dementia
Vol.07No.02(2018), Article ID:85279,23 pages
10.4236/aad.2018.72002
Christopher Busby
Environmental Research SIA, 1117 Latvian Academy of Sciences, Riga, Latvija

Copyright © 2018 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: February 23, 2018; Accepted: June 11, 2018; Published: June 14, 2018
ABSTRACT
The effect of ionising radiation exposure on dementia is approached by applying the causation models of John Stuart Mill and of Sir Austin Bradford Hill to mechanism and epidemiological evidence. Since ionizing radiation is known to kill brain cells in laboratory culture and to affect hippocampal neurogenesis in animal experiments at modest doses, it is reasonable to assume that exposure to radiation must affect neurological integrity and hence dementia rates in those who are exposed. There is persuasive evidence from the epidemiological studies of a large cohort of female nuclear workers that ionising radiation exposure is associated with significant low dose region dose-dependent increases in rates of dementia. Using results from these studies, the Probability of Causation approach (PC), conventionally employed for assessing cancer risk following radiation exposure, is extended to dementia to find a risk coefficient for all ages of 60 per Sievert cumulative exposure over the range 0 - 100 mSv. The finding suggests that natural background external exposures to ionizing radiation are partly responsible for the development of dementia in human populations. A simple general model for dementia is proposed.
Keywords:
Radiation, Alzheimer, Dementia

1. Introduction
Discussion of the health effects of ionising radiation (IR) has, until recently, focused on cancer and genetic damage as assessed largely through the concept of “absorbed dose” and rates of illness and congenital effects revealed by the Japanese Life Span Study (LSS) of the survivors of the Hiroshima and Nagasaki bombs. Indeed, the currently accepted cancer risk and genetic risk per unit dose applied by governments, following advice from radiation risk agencies like the International Commission on Radiological Protection (ICRP), is largely based on the LSS [1]. The conclusions and the safety of the LSS studies will be revisited here. However, it is increasingly clear that IR exposures cause heart disease, and a wide range of other conditions. Indeed, evidence has accumulated to the effect that IR has an impact on most organs and systems of the body through what can be broadly described as “premature ageing” [2]. One of the most-age related and serious conditions increasingly found in human populations is dementia. Dementia, is now the 4th leading cause of death in the USA, the leading component of dementia being Alzheimers disease [3]. There is now considerable evidence that exposure to ionizing radiation can lead to the development of Alzheimers disease and to dementia [4]. Evidence includes results from high dose radiation therapy for brain tumours [5] [6] [7] but also from low dose exposures from e.g. CAT scans and from studies of nuclear workers which will be discussed below. The issue of causation, and the risk of dementia from IR are approached here through a consideration of mechanism, laboratory and epidemiological evidence, using the philosophy of causation as presented by the classic work of Mill [8] and Bradford Hill [9].
2. Causation
Epidemiology of IR and health, presented by risk agencies, rarely discusses the philosophical basis on which their conclusions are drawn, even though the mathematical analyses employed to reduce the evidence to risk coefficients can be impressively complex and daunting, which confers a kind of spurious credibility to the conclusions. The attempts to torture mathematical risk functions to fit the data of the LSS studies may be seen in the approach of the US BEIR VII report [10]. The ICRP is more modest in its approaches and generally relies upon assessing risk through the assumption of a linear no threshold (LNT) function for dose response [1] in which doubling the dose doubles the risk over the whole range of doses from zero to those which cause the rapid death of the individual. This approach continues to be applied despite the clearly complex nature of data which generally shows greater proportionate risk at low doses relative to high doses for almost every type of cancer [11] and the clear impossibility of a linear effect when at the high doses there is death of both tissue and individual. The genetic effects of IR are assumed to be absent in humans, despite animal studies showing quite clearly that they exist. This finding from the LSS study, which has implications for the dementia discussion, will be revisited also. It was peculiar that the finding of absence of genetic risk did not cause the LSS epidemiologists to look more closely at their protocols, assumptions and activities.
Before turning to IR and dementia it may be of value to begin with a brief outline of the approach taken here to assess causation. The issue of causation and evidence was famously first examined some 150 years ago by the philosopher John Stuart Mill and laid out in what has become known as Mill’s Canons [8]. They can usefully be applied to health effects and indeed were, in the 1960s, by British epidemiologist Bradford Hill, whose approach to causation in environmental health is now the classic model [9].
Mill gave five tests for causation. These were:
1) The Canon of Agreement, which states that whatever there is in common between the antecedent conditions of a phenomenon can be supposed to be the cause, or related to the cause, of the phenomenon;
2) The Canon of Difference, which states that the difference in the conditions under which an effect occurs and those under which it does not must be the cause or related to the cause of that effect;
3) The Principle of Accumulation, which states that scientific knowledge grows additively by the discovery of independent laws;
4) The Principle of Instance Confirmation, that the degree of belief in the truth of a law is proportional to the number of favourable instances of the law;
5) Plausibility of a mechanism.
These are the basic methods of science [12].
The application of this approach to causation in the field of epidemiology was carried forward by Bradford Hill in the 1950s. Hill’s influential book Principles of Medical Statistics described what are now termed “Hill’s Canons” and are taught to all doctors and epidemiologists. However, in the case of IR and health, they are rarely applied. They are listed here in Table 1.
The general application of these questions to the issue of IR and health was made in the reports of the European Committee on Radiation Risk (RCRR) [13]. Here, the system will be applied to IR and dementia.
3. Mechanism
3.1. Indirect Link through Ageing
The issue of mechanism in the case of Alzheimers dementia was reviewed recently by Begum et al. [5] who concluded that since dementia was strongly associated with ageing, and IR also was known to increase the rate of ageing through the generation of Reactive Oxygen Species (ROS), there was a logical link between IR exposure and dementia. Significant evidence exists from the after-effects of radiation therapy suggesting that even low doses of IR, such as those from CAT scans, can trigger mechanisms associated with cognitive dysfunction such as that seen in normal ageing [14] [15]. Alzheimer’s disease (AD), an irreversible neurodegenerative condition, accounts for up to 80% of dementias [16] [17]. The pathology involves the accumulation of neuritic plaques and neural fibre tangles (NFT). The main component of the plaques, an accumulation of an amyloid beta peptide derived from the cleavage of an amyloid precursor protein (APP), is associated with inflammation [18] , oxidative stress [19] [20] , NFT formation [21] , neuron loss [22] and the overall effect is AD-related cognitive impairment [23]. The idea that ROS resulted in neurodegeneration is now well established [24]. Oxidative stress produces NFT and neuronal plaques but also affects the neuron populations [25]. Elevated ROS levels cause damage

Table 1. Bradford Hill’s canons for epidemiological causation [9].
to critical components of cellular integrity, DNA, RNA, proteins and lipids [26]. It was recently found that neuronal RNA oxidation was higher in individuals showing mild cognitive impairment than in age matched controls. Increased levels of malondialdehyde (MDA) a compound produced by oxidation of unsaturated lipids, were found in the brains of AD patients [27] and treatment with anti-oxidants reduces AD incidence in patients [28]. Thus, there is considerable evidence that ageing, caused by elevated levels of ROS, leads to dementia. Of course, the biological effects of IR are mainly through the generation of ROS [29] [30] [31]. It is these reactive ions, produced by the interaction of fast electrons with solvent water and other cellular constituents, which react with DNA and produce the genetic and genomic effects which result in cancer and the other well accepted health conditions.
Since IR exposures produce ROS and ROS affects neurodegeneration and dementia, it follows by straightforward logic that IR exposures will increase the rates of dementia through this indirect ageing mechanism. But can this be distinguished from a direct effect?
3.2. Effects of IR on the Brain
It is commonly stated that the brain has low radio-sensitivity. However, it is no longer clear that this is the case, but was an early belief arising from the association between cell replication rate in tissues and radiation sensitivity. There is significant recent evidence arising from research into the effects of cancer radiotherapy, that the brain, or certain regions of the brain involving neuro-generation are in fact, highly radiosensitive. First, the effects of IR cannot be distinguished from the generation of ROS. There is no doubt that isolated nerve cells can be killed by modest doses of IR in vitro. Figure 1 shows the effect of external gamma irradiation of neuroblastoma cells in culture [32]. From the survival curve, 20% of all cells are killed at a dose of about 250 mGy and extrapolation would suggest that 10% of cells would be killed at 50 mGy. This may be compared with human colon HT29 cells in culture where about 2000 mGy is necessary to kill 20% of cells and Human Ovary OVCAR 10 cells which need 1500 mGy to kill 20% [32]. This is because unlike the normal human tissue which is apoptosis absent, human neuroblastoma cells are apoptosis dominant; the cells do not repair and replicate [32]. The loss of neurons in the brain is not reversible. All neuron killing is loss of neuron number in the whole brain. Thus, it is straightforward to argue that neuron death and the associated loss of specific neuron connection over the lifespan of the individual will contribute to cognitive and memory loss and increased brain dysfunction. For example, natural background external gamma doses of about 0.8 mSv per year would give an accumulated brain IR dose of 48 mSv at age 60, the age at which dementia rates begin to increase in human populations. By age 80, where about 1/3rd of the population exhibits cognitive and memory impairment, the cumulated IR dose is approximately 64 mSv and if the cell culture results are applied, there is loss of some 15 to 20% of neurons due to external IR alone. Belka 2001 states that the effectiveness of radiotherapy is frequently limited by the tolerance of CNS structures [33] which is contrary to any belief that the brain is less radiosensitive than other tissues. The authors write that clinical responses to radiotherapy of the brain
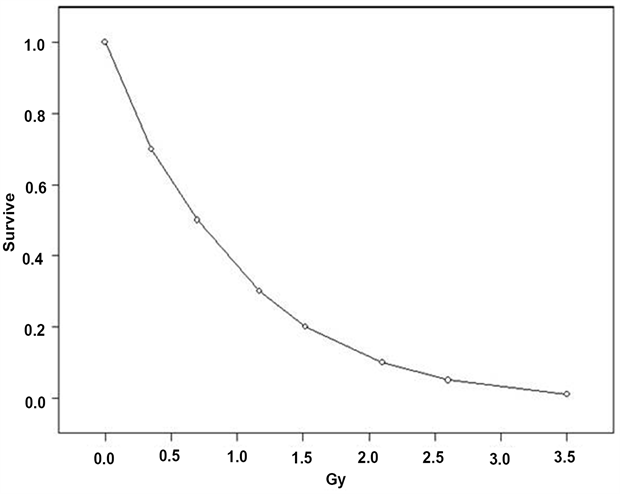
Figure 1. Survival fraction curve for human neuroblastoma cells HX142 and HX 138 irradiated in culture. Re-drawn from Hall 2000 [32].
include a late response involving a diffuse decline in cerebral function with cognitive impairment being the most prominent [33]. Radiotherapy however, involves very large doses, usually tens of Gray. A major problem with attempting to assess IR effects from radiotherapy at these high doses is that individual and pathological data are restricted to cases with pre-existing and extensive clinical problems which would confound any conclusions regarding the effects of IR on a healthy population.
However, there is another important component to the effects of IR on the brain. Recent research identifies neuronal re-capacity as arising from neural precursor cells in the hippocampus. This part of the brain produces multipotent stem/precursors that produce cells that migrate away and produce neurons or glia [34]. One theory is that the cellular and molecular mechanism which underlies radiation induced cognitive impairment involves alterations in hippocampal neurogenesis [35] - [40]. Data from human patients irradiated for brain tumours shows reduction in neurogenic cells [40]. Laboratory studies clearly indicate that these alterations in neurogenesis (and cognitive impairment) involve inflammation [18] [19] [36] [37] [41] [42] [43] [44] [45] and factors related to ROS levels [42] [46].
The question of dose and dose rate necessary to cause such damage arises. A number of studies have found pathological changes and also cognitive effects at various doses [32] [34] [35] [47]. Low doses can lead to cognitive dysfunction without inducing significant morphological effects [46]. In support of the effects on the hippocampal neurogenesis described above, cognitive changes produced by IR are often manifested as hippocampal-dependent functions of learning, memory and spatial information processing [48] [49].
Some reports have suggested that cognitive impairment, including dementia can be observed in up to 50% of long term brain tumour survivors older than 50 years treated by radiotherapy [48] [49].
4. Epidemiology of IR and Dementia
The mechanistic arguments from the rates of dementia with age, together with the association of ageing with ROS and therefore IR-induced ROS generation, support a belief that IR is certainly a component of the cause of dementia in normal human populations exposed only to natural background radiation. Rates of dementia begin to increase only after age 64. The prevalence at ages below 64 are very low, in one study the prevalence rate for ages 45 - 64 was less than 0.02%. For 64 - 69 it was 1.3 and for 70 - 74 it was 2.9, increasing by about double for every five years after that [50]. This suggests that the onset may be a consequence of the overwhelming of a buffer capacity for mentation and memory. This is supported by evidence showing excess risk with smaller brain size [50]. A discussion of how this might happen is presented later in this contribution.
Clearly to assess the risk of IR exposure causation, and following the methodology of Bradford Hill, the ideal experiment would be to obtain dementia incidence or mortality in populations exposed to different doses of IR. This is the method used to obtain risks of cancer and genetic damage from IR. There are obvious sets of populations that can, in principle, be studied to obtain information about dementia and IR exposures. These are:
・ Patients exposed to radiotherapy including radiotherapy of the brain;
・ Hiroshima and Nagasaki A-bomb survivors;
・ Survivors of nuclear accidents;
・ Nuclear workers;
・ Radiologists.
4.1. Post Radiotherapy
Radiotherapy for brain tumours and other cancer and leukemia can deliver very high doses to patients who already have changes in the brain resulting from advanced tumours in the brain or elsewhere in the body. A significant fraction of patients will die a few months or years following the radiation exposure due to the failure to check the development of the cancer. In the Tucker et al. 1989 post radiation study of 52 individuals irradiated for non-Hodgkin lymphoma at St Bartholomew’s Hospital in London, only 12 were alive 10 years later [51]. Accordingly, since the numbers involved in these hospital studies are small to begin with, the expected rate of incidence of dementia in any study group will be generally be too low to expect any result. Table 2 lists a selection of the main studies of dementia and related brain irradiation consequences, symptoms and signs following radiotherapy. Most of the studies could not be termed epidemiological since the ages of each individual being treated and their survival after treatment are not given except as means or medians. An example will suffice to demonstrate the problem of epidemiological power. Let us assume that the radiotherapy causes a ten-fold increase in risk. Let us take a hospital population of 50 radiotherapy patients aged 40 - 50. Being generous, the incidence rate in the normal population is 50 per 100,000. This is increased by the exposure to 500 per 100,000. But since there are only 50 individuals, if we follow all of them for 5 years we would expect only 2500 in 100,000 cases or 0.025 of a case even if they all were still alive at the end of 5 years. In real studies, usually less than 20% survive to follow up. Therefore, the fact that there were no increases in dementia found in the study population, whilst descriptively graphic, really tells us nothing about the effects of the IR exposures. Similar calculations to other patient groups show much the same problem. Therefore, such studies are not useful as a means to quantify the effects of IR although they may be generally informative. What they all do show, however, is measurable and quantifiable effects both in brain morphology and in neurological and neuropsychological assessments which point clearly to measures which are associated with loss of brain function and ultimately to dementia. The issue now is hardly one where there any argument.
What is clear is that there is only one epidemiological series of studies which used a case-control approach, had measured or estimated doses in the low dose range, and which had a large enough population to draw statistically powerful
Table 2. A selection of defining studies of dementia and related neurological effects following radiotherapy and other exposures. N is the number of individuals studied. Note: there are many tens of studies of the neurological outcomes of high dose radiotherapy. All show effects of the kind listed here and all suffer from the small numbers problem addressed in the text.
conclusions. This was the Wilkinson 2000 National Institute of Occupational Health (NIOSH) study which was followed up by Sibley et al. in 2003 and also later in 2006 [57] [58] [59]. All three studies showed an overall significant effect and a highly significant trend in dose response. These studies fulfil the requirements of the Bradford Hill Canons for causation and can be employed to estimate a relative risk for dementia by radiation dose over the low dose region. The result of doing this has interesting implications for normal populations and is carried out below.
4.2. A-Bomb Survivors
One of the problems here is that the rates of dementia increase to levels which are useful for epidemiological power only when the population age is greater than 75. The LSS study has individuals with calculated doses and age at exposure data and reports have apparently shown no significant excess risk of dementia related to dose [63] [64] [65]. The investigation of dementia was, however, restricted to a much smaller cohort (2200) of over 60 s recruited in 1950, the Radiation Effects Research Foundation Adult Health Study cohort, the AHS. The finding was that there was no radiation dose-related effect although the prevalence in the cohort was much higher than in Japan. This raises the issue of the safety of the LSS control group and the LSS studies in general. It was recently pointed out that the removal in 1973 of the unexposed control group which defined the baseline for the radiation cancer studies makes the results of the LSS epidemiology unsafe [66]. Exposures to all three “dose groups” used to obtain cancer risk coefficients included fallout from the post detonation “black rain”. Studies of cancer rates using nearby prefectures as controls were carried out in 2006 and showed significant effects in all dose groups [67]. There are other epidemiological problems with the LSS study [13] [68].
4.3. Chernobyl
Survivors of nuclear accidents, in particular the Chernobyl accident, seem to show significant cognitive deficit effects in a number of studies from the Ukraine and Belarus e.g. [60] but it is difficult to filter out dementia and even more difficult to assess the individual doses, which were mostly internal. However, what we can say is that overall internal doses as calculated by the methodology of the ICRP were all below 20 mSv. At these calculated doses, according to current scientific belief, no effects of any kind should occur. However, this seems to be changing: genetic effects in birth outcomes have been reported in Chernobyl contaminated areas where the doses to populations have been lower than 2 mSv, conventionally assessed. These findings raise the issue of the safety of the ICRP methodology for internal exposures and indeed the calculation and assessment of internal radionuclide doses is currently an area of debate [13] [30] [68]. What is required here is a population where a reasonable measure of an excess radiation exposure dose can be made relative to a normal population exposed only to natural background.
5. Nuclear Worker Cohort
5.1. Wilkinson et al. 2000
Wilkinson et al. 2000 [57] examined causes of death in 67,976 female nuclear workers from the nuclear weapons industry. Although Standardized Mortality Ratios (SMR) for all causes combined were found to be less (69 for badged and 78 for unbadged) than the national population, the SMR for deaths from mental disorders was unusually high at 147. The national population SMR is, of course, 100. Therefore, the risk ratio for the nuclear workers for dying from mental disorders was 1.47, and they had a 47% excess risk of dying from mental disorders than the national population, even though, owing to a “healthy worker effect”, their risk for all causes of deaths was about 25% lower. This unexpected finding led to a larger study which was funded by NIOSH and specifically examined deaths from dementia. The SMR was based upon 166 deaths from mental disorders including 91 from dementia.
Therefore, since 91 deaths represents a 47% excess over the expected number in a national matched population, the expected number of deaths from dementia in this population is 62 (91/1.47).This represents a highly statistically significant result. Mantel Haenszel Chi-square = 5.49 with 95% confidence intervals of 1.06 < Odds Ratio < 2.04; p = 0.019.
5.2. Sibley et al. 2003
The initial finding was then followed up in a further study of the 67,976 female nuclear workers by employing a nested case-control study which looked at the 91 dementia deaths [58]. Because estimates of exposure were only available for 14 cases and 154 controls, the methodology focused on 14 individuals who had measured doses and compared their doses to 154 controls. The effects of internal exposures were not examined since data was not available. Case control studies are generally accepted by epidemiologists as being the gold standard of investigation. Results are given in Table 3 and Figure 2.
From data given in the paper it is a simple matter to calculate the mean lifetime dose to the cases as 20 mSv. Thus, taking the overall OR as 2.23, the Excess Relative Risk ERR (as OR-1) is 1.23 per 20 mSv external dose. A linear extrapolation (such as that employed by the ICRP and other risk agencies for cancer) therefore gives an ERR per Sievert of 1.23 × 1000/20 = 61.5.
This method is exactly that employed to obtain Excess Relative Risk for cancer and leukemia from the Odds Ratio, SMRs and SIRs obtained from external exposure studies like that of the Japanese A-Bomb Lifespan Study (LSS) [1] [10].
The authors of this study conceded that the numbers of cases that they employed for the dose response study was small and proposed that a larger study be carried out as a follow-up. This was done and the number increased to 34. The study is discussed below. It should be borne in mind that, overall, the risk of dying from dementia was significantly greater in the overall nuclear worker study of Wilkinson et al. 2000 where there were 91 deaths, most of which were of those with no radiation doses recorded. This did not, of course, mean that they were not exposed either externally or internally from the higher levels of contamination found at the weapons development sites where they worked. In the current calculation, the internal exposures must necessarily be ignored.
Table 3. Odds Ratios for deaths from dementia in monitored female nuclear workers nested by lifetime doses and maximum annual doses; from Sibley et al. 2003 [58].
*Statistically significant at the p < 0.05 (95%) level; **at the p < 0.01 (99%) level.
5.3. Sibley et al. 2006
Sibley et al. 2006 [59] was a follow-up to Sibley et al. 2003 [58] in which the same population was employed but the definition of dementia in the exposed group was extended to death certificates where dementia was listed as an existing condition or co-cause. This increased the number of cases for which doses were recorded by 20, increasing the total number of monitored individuals from 14 to 34. This increased the statistical power of the study. The results reported in the paper were:
After adjusting for age first monitored and number of years monitored, a relative risk of 1.1 (95% CI = 1.0 - 1.3) per 10 milliSieverts (mSv) was observed for cumulative dose. When 3 cumulative dose categories (0.0 - 9.9 mSV, 10 - 24.9 mSv, and 25+ mSv were employed, an Odds Ratio (OR) of 7.6 (95% CI = 2.3 - 25.0) was observed for the highest relative to the lowest dose category. An OR of 3.0 (95% CI = 1.3 - 7.2) for cumulative doses 10 - 24.9 mSv relative to 0.0 to 9.9 mSv was also observed.
It is clear that a highly statistically significant OR of 3.0 was found for the test of highest to lowest dose category. Employing the same approach, this gives an ERR of 2.0 per 15 mSv (the mean difference of highest to lowest dose) or an ERR per Sievert of 133, about twice that obtained from the Sibley 2003 study. From the construction of the study group in Sibley 2006, we can say that this ERR is larger because it is for dementia existing at death rather than dementia as a cause of death. It is therefore closer to a measure of incidence rather than mortality.
6. Probability of Causation
Excess Relative Risk (ERR) is the excess risk found in an exposed group relative to that which it would have been had the group had the characteristic and illness
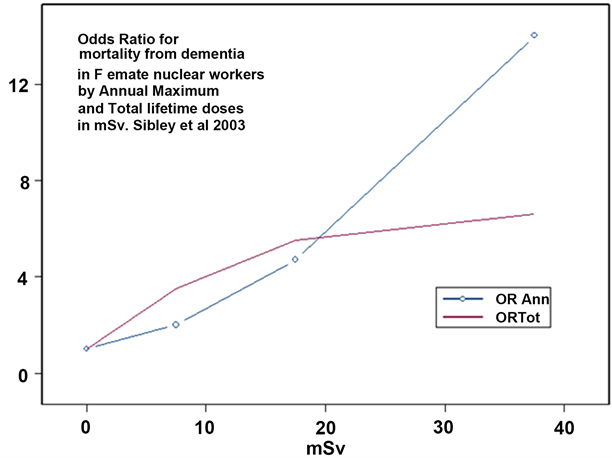
Figure 2. Odds Ratios (OR) for deaths from dementia in female nuclear workers by maximum annual dose and total lifetime dose. Individual points are not statistically significant but overall for both maximum annual and total lifetime doses. Results are statistically significant (p < 0.05) and the linear trend in proportions is also statistically significant (p < 0.006).
rates of the unexposed reference population, usually the national population [10]. If a comparative study shows a Relative Risk of RR as a fraction of the rate in the comparison control population (in the case of the NIOSH study 1.47) and the ERR is 0.47, that is a 47% increase in the rate relative to the comparison population since the RR in the comparison population is, of course, 1.00. The derivation of Probability of Causation (PC) is a straightforward and standard epidemiological process and is employed by the National Institute of Occupational Safety and Health (NIOSH) in its radioecological program IREP to determine PC for radiation induced cancers and leukemias [10] [69]. The process is simply to decide on the Excess Relative Risk (ERR) per Sievert for the cancer of interest, obtained from radiation cancer epidemiology, and then to apply the equation:
(1)
For the cumulative radiation exposure of the average individual, the Excess Relative Risk per Sievert obtained from the Sibley et al. 2003 study of nuclear workers is 61.5.
For a mean external natural background of 0.7 mSv/year the mean dose accumulated between age 20 and age 65 is 28 mSv and
(2)
And the probability of causation for death from radiation induced dementia is:
(3)
This supports the speculation that background radiation exposure may be a major cause of dementia. By age 80 the accumulated dose will be 42 mSv and the PC increases to 72%. One interesting aspect of natural background exposure is that a major component of this is from the radioactive noble gas Radon. It is an interesting fact that Radon, and other noble gases, (like the Krypron-85 emitted in significant quantities by nuclear fission) have about 8-fold more solubility in chemically-defined fats than in water [70]. Thus, the hydrophobic tissue like the myelin that makes up the neural connections in brain and other nervous system tissue would be predicted on a physico-chemical basis to preferentially concentrate Radon and other noble gases. Doses from Radon depend on location and on adequate ventilation in homes affected by releases from the ground and from certain building materials.
7. Discussion
The question being asked here is, does ionizing radiation exposure cause or contribute to the development of dementia? And if so, can we quantify this in some way? We know that ionizing radiation exposure causes biological damage and from the studies discussed and cited above we can see that IR has profound and objectively measurable effects on brain tissue and function. Further, biological damage must affect the ageing process and so we can be sure that ionizing radiation exposure increases the rate of ageing; indeed, studies have been cited that demonstrate this. Dementia is a function of ageing, increasing rapidly from age 65.
Returning to the issue of causation, the requirements of Mill and Bradford Hill can be applied to the evidence reviewed and discussed here.
7.1. Mill and Causation
Referring to Section 2, the Mill Canon of Agreement is satisfied from all the studies cited. All studies of IR exposures discussed show damage to the brain, but particularly the case controls studies of the Wilkinson NIOSH cohort. The Sibley studies specifically address the Canon of Difference since that is the philosophical basis of the case-control study method, which is designed to directly compare situations in which the causal agent is present and where it is absent. The Principle of Accumulation is more difficult to apply here specifically to dementia, but evidence that IR causes a range of illnesses may be included in a discussion of this principle. The Principle of Instance confirmation can certainly be applied to the general brain damaging effects of IR for which there is now overwhelming evidence. However, for the low dose region, we only have the nuclear worker cohort studies. As to Plausibility of Mechanism, there is no shortage of plausible mechanisms available and discussed above for brain damage and eventual dementia following IR exposure. However, the question of the different effects of very high dose and low dose exposures on dementia invites some discussion. This follows in Section 7.3.
7.2. Bradford Hill and Causation
Bradford Hill requires that a causation finding is statistically significant. The nuclear worker studies show a highly statistically significant excess dementia risk, initially with a very large sample, some 69,000 women and 91 deaths when 62 are predicted from the national data (Chi-square 5.49 with 95% confidence intervals of 1.06 < Odds Ratio < 2.04; p = 0.019. These workers would have had a greater mean cumulative dose from external and internal IR exposures and so we cannot say exactly what the mean overall doses were. Furthermore, these were healthy workers: their SMR for all causes of death was about 0.7 and thus the real excess SMR for dementia would have been closer to 2. If we assume a mean dose of 10 mSv for the whole group, the excess risk coefficient (ERR) per Sievert over the low dose range is 100. The restriction of the study group to those with recorded doses also results in a statistically significant excess risk (Table 3). The Strength of Association is sufficient to argue that this is a serious problem that is real. Consistency cannot be found for dementia and IR in specific studies beyond the nuclear workers. The A-Bomb study results are suspect because of the removal of the true control group, but further studies of dementia in the LSS overall cohorts could be carried out using neighbouring prefectures. Regarding specificity and reversibility, permanent brain damage following high doses are accepted, and of course these are not reversible. The relationship in time requirement is fulfilled. There is a statistically significant biological gradient in the nuclear worker studies (p = 0.006). There is biological plausibility through a number of mechanisms described above. Finally, can there be an alternative explanation? The answer, following all that has been found in the many studies of IR and brain damage, is no. There is nothing that ties together nuclear workers, Chernobyl victims and cranial irradiation patients but IR. It is time to accept that IR exposure increases the incidence of dementia through a number of mechanisms, and that the effect exists at low doses of external radiation, and probably also at low doses of internal exposures.
7.3. Towards a General Mechanistic Model for Explaining the Observations
The Persistence of Memory Model
The evidence presented above from the literature shows that changes occurring in the brain that are associated with changes seen in dementia may brought about at IR exposure levels which are relatively low [14]. These are measured changes to genetic markers in the hippocampus. But at the same time, several studies have shown that the extremely high doses to the brain, following radiotherapy, seem to induce memory problems in the short term but perhaps do not seem to have the proportionate long-term effect on memory that might be expected given the low dose effects. Of course, as has been pointed out above, the epidemiology of those exposed to very high doses is highly constrained by statistical power and by confounding causes of death, since these are always individuals suffering from serious diseases. Nevertheless, the issue of very high dose and low dose effects, which may operate through different mechanisms is an intriguing one and will be addressed here.
There are clearly two main components of catastrophic memory loss. One is the straightforward rate of death of neurons and their infrastructural connections. The other is the potential replacement of neurons through the pluripotential cells provided by the hippocampus. For this, the components of a mathematical model may be selected and a model constructed. A dementia model which incorporates the observations is outlined in Figure 3. It is proposed that the condition arises out of an age-related and radiation-related (not independent) destruction of neurons and associated memory at a rate which exceeds replacement of memory through existing spare neuronal capacity and the production of new neuronal capacity through the hippocampus. This mechanism and its kinetic equations will be presented and more fully elaborated in a separate contribution. However, the basic discursive approach, ignoring variation in kinetic rates, is as follows and is illustrated in Figure 3.
If the uniform rate of loss of neurons in the memory structures of the two brain hemispheres is c then ct is the number of neurons lost in anytime t after birth. Then let bt, in the same way, represent the number of new neurons created from pluripotential cells from the hippocampus and which are available for new connections to replicate memory structures.
Then let a be the total number of memory neurons in the brain at t = 0.
We assume that each memory structure requires a mean number of N neurons to be connected. So, there are a/N potentially available memory structures at t = 0.
Then −ct/N represents memory loss, the number of memories lost per unit time.
bt/N represents potential copying of memories to new neurons provided by the hippocampus and thus the recovery of memories through this mechanism.
Then dementia onset occurs when there is no further capacity. This is at time T when the overall memory capacity is overwhelmed:
(4)
Clearly this will be affected by (T will change):
a) The brain size to begin with at t = 0 (genetics, cranial capacity, head volume); N The number of memories used or required (intellectuals vs. farmers).
b) The efficiency and development of the hippocampus (stress, radiation, ROS, genetics).
c) The rate of damage or death of the neuronal population (radiation, ROS, trauma).
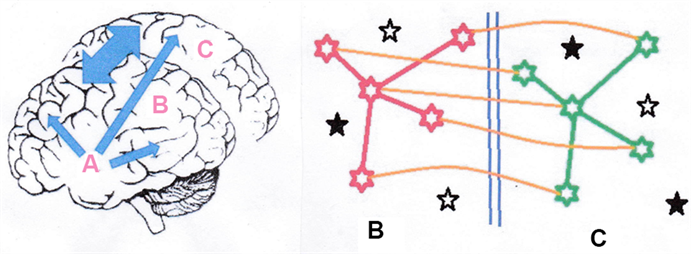
Figure 3. Persistence of Memory. The figure on the right is a representation of a schematic memory image structure consisting of a 5-neuron pattern which is copied back and forth between the stylized brain hemispheres B (red) and C (green) and also shown as double ended arrow in the figure on the left. The corpus callosum is indicated as blue vertical lines in the right-hand figure. The memory is sustained since any damage to a neuron or even the whole memory image structure in hemisphere B shown in the figure on the left can be rebuilt from information still existing in hemisphere C and vice versa through the (orange) connections between the hemispheres. These new links can also be transferred in the model to create connections to entirely new neurons. There should always be capacity as long as there exists viable but unconnected neurons (the full black stars) so new identical memory structures can be created. The total capacity is continuously supported by new pluripotential cells (open black stars) provided by the hippocampus A in the left figure (blue arrows). Failure of this support system A will result in loss of free memory capacity and early onset of dementia. Through this mechanism, memory can survive major trauma so long as the damage is asymmetrical, e.g. a shotgun blast to half of the head. For the treatment and discussion see text.
The model presented in Figure 3 assumes that memory is shared between the two hemispheres, and that the reason for the evolutionary development of two hemispheres is that memory is stored as a matrix of connections between neurons. In a system like the brain, where, unlike other organs in the body, cells do not repair and replicate to sustain tissue integrity, it is not the individual cells which are important, but the system of interconnecting neurons that is important; it is the connections which represent the memory structures (Figure 3). Clearly, then, as with the double helical conjugate system evolved to maintain DNA information integrity, there has to be a copy made by the brain somewhere which can be used to maintain the persistence of memory. This has to be the 2-hemisphere mirror system. Then when part of a specific memory is destroyed in one hemisphere, that memory can be re-instated somewhere in the same hemisphere through new connections made from the conjugate hemisphere. This explains the puzzling observation of memory recovery after massive trauma. So long as sufficient components of memory remain in the non-traumatised hemisphere, or even in parts of the 2 hemispheres, the memory will be re-constructable, will persist and in time will recover through the copying back from the undamaged hemisphere(s). Indeed, temporary memory loss which is regained in a few months is seen in those exposed to very high radiation doses. Also, in a system where it is the connections rather than the neurons that define memory and where neurons do not divide (and indeed there would be no point) there is a requirement for new neurons to be produced, and it seems that such a system exists in the hippocampus.
The possibility of this repair of memory will assume that there is spare neuronal capacity and this raises the issue of such capacity. In the model, there is assumed to be baseline number of neurons, usually far more (we assume) than will be needed in an average lifetime, though this may distinguish between those who require large amounts of memory (e.g. intellectuals) and those who do not (e.g. farmers). In addition, there is the replacement system through the activity of the hippocampus, which is likely to be regulated in some way. The failure of memory occurs, as is seen in Equation (4) when the total capacity of the available memory becomes zero as a result of a combination of neuronal destruction by ageing and therefore radiation, with insufficient or zero production of new neurons from the hippocampus.
The effects of low doses of IR on the hippocampus have been measured: they are mediated not by damage, but by induced alteration in activity [14]. These changes must be regulatory, and their purpose is not clear. However, they are seen, and have similar characteristics to changes seen in dementia patients, and so it is easy to argue that they are associated with a reduction in the production of the pluripotential cells necessary to compensate for the rate of loss of neuronal memory.
8. Conclusion
There is now sufficient mechanistic and epidemiological evidence to accept that exposure to external ionising radiation at low doses < 100 mSv is a risk factor for dementia. A linear risk coefficient of 60 per Sievert for dementia mortality over the range 0 - 100 mSv is deduced from nuclear worker studies. On this basis, lifetime cumulative external ionizing radiation dose is a predictor for development of dementia in human populations and a mechanism which predicts and explains the main observations has been proposed.
Acknowledgements
I am grateful for the late Salvador Dali’s famous and strange surrealist painting entitled “The Persistence of Memory” which I stole for the dementia model I have proposed and will more fully develop in a separate contribution. Interestingly, the central fish like object expiring on the on the beach in Dali’s painting could well be taken as a dream depiction of the poor little hippocampus, the hero of this story of memory and it increasingly seems, our mental equilibrium. I am grateful to Dr Fred Mettler for drawing my attention to the issue, and particularly the evidence from very high dose radiotherapy studies, which set me on the road to examining these interesting and important issues.
Cite this paper
Busby, C. (2018) A Risk Coefficient for Radiation-Induced Dementia. Advances in Alzheimer’s Disease, 7, 13-35. https://doi.org/10.4236/aad.2018.72002
References
- 1. ICRP (2007) The 2007 Recommendations of the International Commission on Radiological Protection. ICRP Publication 103. Annals of the ICRP, 37, 1-332.
- 2. Richardson, R.B. (2009) Ionizing Radiation and Aging: Rejuvenating an Old Idea. Aging, 1, 887-902. https://doi.org/10.18632/aging.100081
- 3. Hebert, L.E., Scherr, P.A., Bienias, J.L., et al. (2003) Alzheimer Disease in the US Population: Prevalence Estimates Using the 2000 Census. Archives of Neurology, 60, 1119-1122. https://doi.org/10.1001/archneur.60.8.1119
- 4. Becker, N., Liebermann, D., Wesch, H., et al. (2008) Mortality among Thorotrast-Exposed Patients and an Unexposed Comparison Group in the German Thorotrast Study. European Journal of Cancer, 44, 1259-1268. https://doi.org/10.1016/j.ejca.2008.02.050
- 5. Begum, N., Wang, B., Mori, M. and Vares, G. (2012) Does Ionizing Radiation Influence Alzheimer’s Disease Risk? Journal of Radiation Research, 53, 815-822. https://doi.org/10.1093/jrr/rrs036
- 6. Asai, A., Matsutani, M., Kohno, T., et al. (1989) Subacute Brain Atrophy after Radiation Therapy for Malignant Brain Tumor. Cancer, 63, 1962-1974. https://doi.org/10.1002/1097-0142(19890515)63:10<1962::AID-CNCR2820631016>3.0.CO;2-V
- 7. Imperato, J.P., Paleologos, N.A. and Vick, N.A. (1990) Effects of Treatment on Long-Term Survivors with Malignant Astrocytomas. Annals of Neurology, 28, 818-822. https://doi.org/10.1002/ana.410280614
- 8. Mill, J.S. (1879) A System of Logic. Longmans, Green & Co., London.
- 9. Bradford Hill, A. (1966) Principles of Medical Statistics. The Lancet Limited, London.
- 10. BEIR VII (2007) The Health Risks of Exposure to Low Levels of Ionizing Radiation. National Academy of Sciences, Washington DC.
- 11. Cardis, E., Vrijheid, M., Blettner, M., et al. (2007) The 15-Country Collaborative Study of Cancer Risk among Radiation Workers in the Nuclear Industry: Estimates of Radiation-Related Cancer Risks. Radiation Research, 167, 396-416. https://doi.org/10.1667/RR0553.1
- 12. Harre, R. (1985) The Philosophies of Science. Oxford University Press, Oxford.
- 13. Busby, C., Yablolov, A.V., Schmitz Feuerhake, I., Bertell, R. and Scott Cato, M. (2010) ECRR 2010: The 2010 Recommendations of the European Committee on Radiation Risk. The Health Effects of Ionizing Radiation at Low Doses and Low Dose Rates. ECRR, Brussels.
- 14. Lowe, X.R., Bhattacharya, S., Marchetti, F., et al. (2009) Early Brain Response to Low-Dose Radiation Exposure Involves Molecular Networks and Pathways Associated with Cognitive Functions, Advanced Aging and Alzheimer’s Disease. Radiation Research, 171, 53-65. https://doi.org/10.1667/RR1389.1
- 15. Kempf. S.J., Janik, D., Barjaktarovic, Z., Braga-Tanaka III, I., Tanaka, S., Neff, F., Saran, A., Larsen. M.R. and Tapio, S. (2016) Chronic Low-Dose-Rate Ionising Radiation Affects the Hippocampal Phosphoproteome in the ApoE-/- Alzheimer’s Mouse Model. Oncotarget, 7, 71817-71832.
- 16. Demarin, V., Zavoreo, I., Kes, V.B., et al. (2011) Biomarkers in Alzheimer’s Disease. Clinical Chemistry and Laboratory Medicine, 49, 773-778. https://doi.org/10.1515/CCLM.2011.139
- 17. Schaeffer, E.L., Figueiro, M. and Gattaz, W.F. (2011) Insights into Alzheimer Disease Pathogenesis from Studies in Transgenic Animal Models. Clinics, 66, 45-54.https://doi.org/10.1590/S1807-59322011001300006
- 18. Wyss-Coray, T. and Rogers, J. (2012) Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harbor Perspectives in Medicine, 2, a006346. https://doi.org/10.1101/cshperspect.a006346
- 19. Nunomura, A., Perry, G., Aliev, G., et al. (2001) Oxidative Damage Is the Earliest Event in Alzheimer Disease. Journal of Neuropathology & Experimental Neurology, 60, 759-767. https://doi.org/10.1093/jnen/60.8.759
- 20. Smith, M.A., Hirai, K., Hsiao, K., et al. (1998) Amyloid-β Deposition in Alzheimer Transgenic Mice Is Associated with Oxidative Stress. Journal of Neurochemistry, 70, 2212-2215. https://doi.org/10.1046/j.1471-4159.1998.70052212.x
- 21. Jucker, M. and Walker, L.C. (2011) Pathogenic Protein Seeding in Alzheimer Disease and Other Neurodegenerative Disorders. Annals of Neurology, 70, 532-540. https://doi.org/10.1002/ana.22615
- 22. Calissano, P., Matrone, C. and Amadoro, G. (2009) Apoptosis and in Vitro Alzheimer’s Disease Neuronal Models. Communicative & Integrative Biology, 2, 163-169. https://doi.org/10.4161/cib.7704
- 23. Patrias, L.M., Klaver, A.C., Coffey, M.P., et al. (2011) Effects of External Beam Radiation on in Vitro Formation of Abeta1-42 Fibrils and Preformed Fibrils. Radiation Research, 175, 375-381. https://doi.org/10.1667/RR2448.1
- 24. Valko, M., Leibfritz, D., Moncol, J., et al. (2007) Free Radicals and Antioxidants in Normal Physiological Functions and Human Disease. The International Journal of Biochemistry & Cell Biology, 39, 44-84. https://doi.org/10.1016/j.biocel.2006.07.001
- 25. Hirai, K., Aliev, G., Nunomura, A., et al. (2001) Mitochondrial Abnormalities in Alzheimer’s Disease. Journal of Neuroscience, 21, 3017-3023. https://doi.org/10.1523/JNEUROSCI.21-09-03017.2001
- 26. Markesbery, W.R. and Lovell, M.A. (2007) Damage to Lipids, Proteins, DNA, and RNA in Mild Cognitive Impairment. Archives of Neurology, 64, 954-956. https://doi.org/10.1001/archneur.64.7.954
- 27. Dalle-Donne, I., Rossi, R., Colombo, R., et al. (2006) Biomarkers of Oxidative Damage in Human Disease. Clinical Chemistry, 52, 601-623. https://doi.org/10.1373/clinchem.2005.061408
- 28. Mancuso, C., Bates, T.E., Butterfield, D.A., et al. (2007) Natural Antioxidants in Alzheimer’s Disease. Expert Opinion on Investigational Drugs, 16, 1921-1931. https://doi.org/10.1517/13543784.16.12.1921
- 29. BEIR V (1990) The Health Effects of Exposure to Low Levels of Ionising Radiation. Committee on Biological Effects of Ionising Radiation, National Academy Press, Washington DC.
- 30. Busby, C. (2013) Aspects of DNA Damage from Internal Radionuclides. In: Chen, C., Ed., New Research Directions in DNA Repair, IntechOpen, 597-637.http://www.intechopen.com/books/new-research-directions-in-dna-repair/aspects-of-dna-damage-from-internal-radionuclides https://doi.org/10.5772/53942
- 31. UNSCEAR (2000) Sources and Effects of Ionising Radiation, Report to the General Assembly, with Scientific Annexes. United Nations, New York.
- 32. Hall Eric, J. (2006) Radiobiology for the Radiologist. Lippincott, Philadelphia.
- 33. Belka, C., Budach, W., Kortmann, R.D., et al. (2001) Radiation Induced CNS Toxicity—Molecular and Cellular Mechanisms. British Journal of Cancer, 85, 1233-1239. https://doi.org/10.1054/bjoc.2001.2100
- 34. Fike, J.R., Rosi, S. and Limoli, C.L. (2009) Neural Precursor Cells and Radiation Sensitivity. Seminars in Radiation Oncology, 19, 122-132. https://doi.org/10.1016/j.semradonc.2008.12.003
- 35. Raber, J., Fan, Y., Matsumori, Y., Liu, Z., Weinstein, P.R., Fike, J.R. and Liu, J. (2004) Irradiation Attenuates Neurogenesis and Exacerbates Ischemia-Induced Deficits. Annals of Neurology, 55, 381-389. https://doi.org/10.1002/ana.10853
- 36. Raber, J., Rola, R., LeFevour, A., Morhardt, D.R., Curley, J., Mizumatsu, S., VandenBerg, S.R. and Fike, JR. (2004) Radiation-Induced Cognitive Impairments Are Associated with Changes in Indicators of Hippocampal Neurogenesis. Radiation Research, 162, 39-47. https://doi.org/10.1667/RR3206
- 37. Rola, R., Raber, J., Rizk, A., Otsuka, S., VandenBerg, S.R., Morhardt, D.R. and Fike, J.R. (2004) Radiation-Induced Impairment of Hippocampal Neurogenesis Is Associated with Cognitive Deficits in Young Mice. Experimental Neurology, 188, 316-330.
- 38. Madsen, T.M., Kristjansen, P.E., Bolwig, T.G. and Wortwein, G. (2003) Arrested Neuronal Proliferation and Impaired Hippocampal Function Following Fractionated Brain Irradiation in the Adult Rat. Neuroscience, 119, 635-642.
- 39. Winocur, G., Wojtowicz, J.M., Sekeres, M., Snyder, J.S. and Wang, S. (2006) Inhibition of Neurogenesis Interferes with Hippocampus-Dependent Memory Function. Hippocampus 16, 296-304. https://doi.org/10.1002/hipo.20163
- 40. Gage, F.H. (2000) Mammalian Neural Stem Cells. Science, 287, 1433-1438. https://doi.org/10.1126/science.287.5457.1433
- 41. Monje, M.L., Vogel, H., Masek, M., Ligon, K.L., Fisher, P.G. and Palmer, T.D. (2007) Impaired Human Hippocampal Neurogenesis after Treatment for Central Nervous System Malignancies. Annals of Neurology, 62, 515-250. https://doi.org/10.1002/ana.21214
- 42. Fike, J.R., Rola, R. and Limoli, C.L. (2007) Radiation Response of Neural Precursor Cells. Neurosurgery Clinics of North America, 18, 115-127. https://doi.org/10.1016/j.nec.2006.10.010
- 43. Mizumatsu, S., Monje, M.L., Morhardt, D.R., Rola, R., Palmer, T.D. and Fike, J.R. (2003) Extreme Sensitivity of Adult Neurogenesis to Low Doses of X-Irradiation. Cancer Research, 63, 4021-4027.
- 44. Monje, M.L., Mizumatsu, S., Fike, J.R. and Palmer, T.D. (2002) Irradiation Induces Neural Precursor-Cell Dysfunction. Nature Medicine, 8, 955-962. https://doi.org/10.1038/nm749
- 45. Monje, M.L., Toda, H. and Palmer, T.D. (2003) Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science, 302, 1760-1765. https://doi.org/10.1126/science.1088417
- 46. Giedzinski, E., Rola, R., Fike, J.R. and Limoli, C.L. (2005) Efficient Production of Reactive Oxygen Species in Neural Precursor Cells after Exposure to 250 MeV Protons. Radiation Research, 164, 540-544. https://doi.org/10.1667/RR3369.1
- 47. Rola, R., Zou, Z., Huang, T.-T., Fishman, K., Baure, J., Rosi, S., Milliken, H., Limoli, C.L. and Fike, J.R. (2007) Lack of Extracellular Superoxide Dismutase (EC-SOD) in the Microenvironment Impacts Radiation-Induced Changes in Neurogenesis. Free Radical Biology and Medicine, 42, 1133-1145. https://doi.org/10.1016/j.freeradbiomed.2007.01.020
- 48. Schultheiss, T.E., Kun, L.E., Ang, K.K., et al. (1995) Radiation Response of the Central Nervous System. International Journal of Radiation Oncology, Biology, Physics, 31, 1093-1112. https://doi.org/10.1016/0360-3016(94)00655-5
- 49. Raber, J. (2010) Unintended Effects of Cranial Irradiation on Cognitive Function. Toxicologic Pathology, 38, 198-202.
- 50. Prince, M., Albanese, E., Guerchet, M. and Prina, M. (2014) World Alzheimer Report 2014. Dementia and Risk Reduction. Alzheimers Disease International, London.
- 51. Tucker, J., Prior, P.F., Green, C.R., et al. (1989) Minimal Neuropsychological Sequelae Following Prophylactic Treatment of the Central Nervous System in Adult Leukemia and Lymphoma. British Journal of Cancer, 60, 775-780. https://doi.org/10.1038/bjc.1989.358
- 52. Grosshans, D.R., Meyers, C.A., Allen, P.K., et al. (2008) Neurocognitive Function in Patients with Small Cell Lung Cancer. Effect of Prophylactic Cranial Irradiation. Cancer, 112, 589-595. https://doi.org/10.1002/cncr.23222
- 53. Peper, M., Steinvorth, S., Schraube, P., et al. (2000) Neurobehavioral Toxicity of Total Body Irradiation: A Follow-Up in Long-Term Survivors. International Journal of Radiation Oncology, Biology, Physics, 46, 303-311. https://doi.org/10.1016/S0360-3016(99)00442-3
- 54. Neider, C., Leicht, A., Motaref, B., et al. (1999) Late Radiation Toxicity after Whole Brain Radiotherapy: The Influence of Antiepileptic Drugs. American Journal of Clinical Oncology: Cancer Clinical Trials, 22, 573-579. https://doi.org/10.1097/00000421-199912000-00007
- 55. Frytak, S., Shaw, J.N., O’Neill, B.P., et al. (1989) Leukencephalopathy in Small Cell Lung Cancer Patients Receiving Prophylactic Cranial Irradiation. American Journal of Clinical Oncology, 12, 27-33. https://doi.org/10.1097/00000421-198902000-00007
- 56. Schulte, P.A., Burnett, C.A., Boeniger, M.S. and Johnson, J. (1996) Neurodegenerative Diseases: Occupational Occurrence and Potential Risk Factors, 1982 through 1991. American Journal of Public Health, 86, 1281-1288. https://doi.org/10.2105/AJPH.86.9.1281
- 57. Wilkinson, G.S., Trieff, N., Graham, R. and Proire, R.L. (2000) Study of Mortality among Female Nuclear Weapons Workers. NIOSH, Washington DC.
- 58. Sibley, R.F., Moscato, B.S., Wilkinson, G.S. and Natarajan, N. (2003) Nested Case-Control Study of External Ionizing Radiation Dose and Mortality from Dementia within a Pooled Cohort of Female Nuclear Weapons Workers. American Journal of Industrial Medicine, 44, 351-538. https://doi.org/10.1002/ajim.10288
- 59. Sibley, R., Wilkinson, G. and Servoss, T. (2006) A Case Control Study of Dementia in Female Nuclear Workers. Epidemiology, 17, S521. https://doi.org/10.1097/00001648-200611001-01402
- 60. Loganovsky, K. (2009) Do Low Doses of Ionizing Radiation Affect the Human Brain? Data Science Journal, 8, BR13-BR35.https://www.researchgate.net/publication/220390293_Do_Low_Doses_of_Ionizing_Radiation_Affect_the_Human_Brain https://doi.org/10.2481/dsj.br-04
- 61. Johnson, B.E., Becker, B., Goff, W.B., et al. (1985) Neurologic, Neuropsychologic and Computed Cranial Tomography Scan Abnormalities in 2- to 10-Year Survivors of Small-Cell Lung Cancer. Journal of Clinical Oncology, 3, 1659-1667. https://doi.org/10.1200/JCO.1985.3.12.1659
- 62. Marazziti, D., Baroni, S., Lombardi, A., et al. (2014) Psychiatric Effects of Ionizing Radiation. The Clinical Neuropsychologist, 11, 61-67.
- 63. Preston, D.L., Shimizu, Y., Pierce, D.A., et al. (2003) Studies of Mortality of Atomic Bomb Survivors. Report 13: Solid Cancer and Noncancer Disease Mortality: 1950-1997. Radiation Research, 160, 381-407. https://doi.org/10.1667/RR3049
- 64. Little, M.P. (2009) Cancer and Non-Cancer Effects in Japanese Atomic Bomb Survivors. Journal of Radiological Protection, 29, A43-A59. https://doi.org/10.1088/0952-4746/29/2A/S04
- 65. Yamada, M., Sasaki, H., Mimori, Y., et al. (1999) Prevalence and Risks of Dementia in the Japanese Population: RERF’s Adult Health Study Hiroshima Subjects. Radiation Effects Research Foundation. Journal of the American Geriatrics Society, 47, 189-195. https://doi.org/10.1111/j.1532-5415.1999.tb04577.x
- 66. Busby, C. (2016) Letter to the Editor on “The Hiroshima/Nagasaki Survivor Studies: Discrepancies between Results and General Perception” by Bernard R Jordan. Genetics, 204, 1627-1629. https://doi.org/10.1534/genetics.116.195339
- 67. Wanatabe, T., Miyao, M., Honda, R. and Yamada, Y. (2008) Hiroshima Survivors Exposed to Very Low Doses of A-Bomb Primary Radiation Showed a High Risk of Cancers. Environmental Health and Preventive Medicine, 13, 264-270. https://doi.org/10.1007/s12199-008-0039-8
- 68. Schmitz-Feuerhake, I., Busby, C. and Pflugbeil, P. (2016) Genetic Radiation Risk: A Neglected Topic in the Low Dose Debate. Environmental Health and Toxicology, 31, e2016001.
- 69. Centre for Disease Control CDC (2014) NIOSH Radiation Dose Reconstruction: Probability of Causation. Atlanta. http://www.cdc.gov/niosh/ocas/pccalc.html
- 70. Clever, H.L. (1979) Krypton, Xenon and Radon. IUPAC Solubility Series, Vol. 2, Pergamon Press, Oxford.
Abbreviations
AD: Alzheimer’s Disease.
AHS: Japanese A-Bomb Adult Health Study cohort.
APP: Amyloid precursor protein.
BEIR: Biological Effects of Ionising Radiation committee of the US National Academy of Sciences.
CAT: Computerised Axial Tomography.
DNA: Deoxyribonucleic acid.
ECRR: European Committee on Radiation Risk.
ERR: Excess Relative Risk: this is 1+RR and represents the excess fractional risk in the study group which can be ascribed to the exposure received by the study group.
ICRP: International Commission on Radiological Protection.
IR: Ionising radiation.
IREP: Ionising Radiation Epidemiological Probability.
LNT: Linear No Threshold. The conventional assumed Dose Response function for the health effects of ionising radiation.
LSS: Life Span Study of the effects of radiation on the Japanese A-Bomb cohorts in Hiroshima and Nagasaki.
MDA: Malondialdehyde.
NFT: Neural Fibre Tangles.
NIOSH: National Institute of Occupational Safety and Health.
OR: Odds Ratio: the ratio of disease rate between two matched populations.
PC: Probability of Causation: the fractional probability that the disease or condition for which a RR has been obtained from a study has been caused by a specific numerical exposure. PC = ERR/(1+ERR); sometimes also expressed as a percentage.
RNA: Ribonucleic acid.
ROS: Reactive Oxygen Species: the highly reactive fragments of water and other materials in the body caused by ionising radiation but also as intermediates in some chemical reactions.
RR: Relative Risk: the ratio of age and sex adjusted disease rate in a study group relative to that in the population including the study group members.
SMR: Standardised Mortality Ratio; the mortality rate relative to the national population rate.
上一篇:Longitudinal Follow-Up of a Po 下一篇:Effects of Syzygium cumini See
最新文章NEWS
- Longitudinal Follow-Up of a Population with MCI: Predictive Value of the MIS Test with Delayed Recal
- Gabapentin for the treatment of behavioral and psychological symptoms of dementia
- Human butyrylcholinesterase knock-out equivalent: Potential to assess role in Alzheimer’s disease
- Letter to the Editor
- Advances in Alzheimer’s disease (AAD): Standing firm at its first anniversary
- Confirmation of the Experimentally-Proven Therapeutic Utility of Madecassoside in an Aβ1-42 Infusion
- Activated Caspase-3 and Neurodegeneration and Synaptic Plasticity in Alzheimer’s Disease
- Knowledge, Attitudes and Beliefs about Dementia in an Urban Xhosa-Speaking Community in South Africa
推荐期刊Tui Jian
- Chinese Journal of Integrative Medicine
- Journal of Genetics and Genomics
- Journal of Bionic Engineering
- Pedosphere
- Chinese Journal of Structural Chemistry
- Nuclear Science and Techniques
- 《传媒》
- 《中学生报》教研周刊
热点文章HOT
- Analyzing Nootropic Effect of Phyllanthus reticulatus Poir. on Cognitive Functions, Brain Antioxidan
- Confirmation of the Experimentally-Proven Therapeutic Utility of Madecassoside in an Aβ1-42 Infusion
- Psychophysiological Effects of a Combination of Sideritis and Bacopa Extract (memoLoges®) in 32 Subj
- Two Cases of Severe Dementia Showing Dramatic Improvement after Denture Placement
- Advances in Alzheimer’s disease (AAD): Standing firm at its first anniversary
- Chronic Administration of Curcuma longa Extract Improves Spatial Memory-Related Learning Ability in
- Activated Caspase-3 and Neurodegeneration and Synaptic Plasticity in Alzheimer’s Disease
- Caring for Individuals with Early-Onset Dementia and Their Family Caregivers: The Perspective